De uma mutação genética a uma doença letal
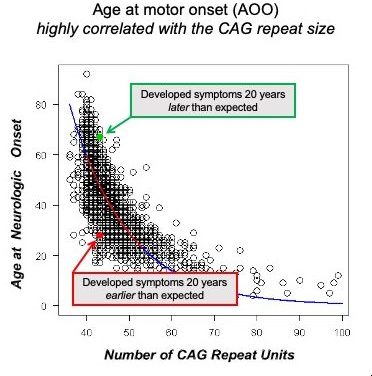
A DH é causada por uma única mutação em uma região do gene HTT, localizada no início do gene (no exon 1), que contém uma sequência repetida composta por repetições CAG; na faixa normal, as pessoas têm entre 17 e 22 repetições em média; em indivíduos com DH, uma expansão superior a 39 conduz inevitavelmente à doença. Quanto maior o número de repetições, normalmente, mais cedo o indivíduo afetado começa a apresentar sintomas visíveis da doença.
Isso significa que todas as pessoas com DH têm o mesmo tipo de mutação em apenas um gene, tornando a DH uma doença incomum. a maioria das outras doenças neurodegenerativas comuns tem muitas origens diferentes, e apenas entre 2-5% de todos os casos de Alzheimer, Parkinson ou ALS têm uma base genética.
Muita pesquisa tem sido dedicada a descobrir como a mutação no gene HTT leva à DH, e muitos mecanismos celulares potenciais foram identificados. O impacto da mutação tem consequências devastadoras para muitas células cerebrais, e a teoria diz que, se pudermos entender como isso acontece, poderemos desenvolver terapias para tratar a doença. No entanto, esses esforços têm sido amplamente infrutíferos até agora. Algumas terapias sintomáticas, direcionadas a um ou outro mecanismo que se pensa estar errado em humanos afetados pela DH, estão em desenvolvimento (ver Tabela abaixo).
Outra abordagem para tratar a DH é visar sintomas específicos da doença que se pensa surgirem de alterações em circuitos cerebrais específicos. O HTT mutante leva à perda de células e fibras cerebrais em circuitos específicos do cérebro, o que explica em parte muitos dos sintomas da doença. Portanto, fortalecer ou restaurar esses circuitos pode oferecer algum benefício. No caso da DH, muitos circuitos são afetados, mas os principais envolvem a conexão entre várias áreas corticais e os gânglios da base, embora outras alterações sejam conhecidas no cerebelo e no hipotálamo.
Esta é a abordagem 'tradicional' adotada para terapias sintomáticas em todos os distúrbios degenerativos e que tem sido eficaz no controle de alguns sintomas. Por exemplo, levou ao desenvolvimento da tetrabenazina para controle do movimento colérico e ao uso de medicamentos antipsicóticos visando o sistema dopaminérgico para irritabilidade, por exemplo. No entanto, neste momento, carecemos de terapias que visem os sintomas mais debilitantes da DH – alterações cognitivas e apatia.
Nesta classe de agentes terapêuticos pode caber o programa atual mais avançado para o tratamento de HD, envolvendo o atual ensaio de Fase 3 (agora recrutamento) de pridopidina, também conhecido formalmente como ACR16, sendo perseguido pela empresa Prilenia. ACR16 foi anteriormente desenvolvido pela Teva.
A pridopidina foi investigada como tratamento para a DH em três ensaios clínicos randomizados, duplo-cegos e controlados por placebo: HART, MermaiHD e PRIDE-HD. Os estudos iniciais na DH centraram-se nos efeitos da pridopidina nos parâmetros motores, sob a hipótese de que a pridopidina tinha um efeito no controlo dopaminérgico do movimento. De fato, um pequeno efeito nos endpoints motores foi observado nos estudos HART e MermaiHD, embora as pessoas também tenham visto um pequeno efeito na escala de capacidade funcional total (TFC) e estenderam o estudo de fase 2 em uma modificação aberta para 52 semanas.
No estudo PRIDE-HD, a dosagem de pridopidina demonstrou um efeito pequeno mas benéfico no TFC e este efeito pareceu ser mais pronunciado para os participantes iniciais da DH, o que levou os investigadores a alargar estas observações num estudo de Fase 3, PROOF-HD, actualmente a incluir e usando a pontuação UHDRS-TFC como o endpoint primário.
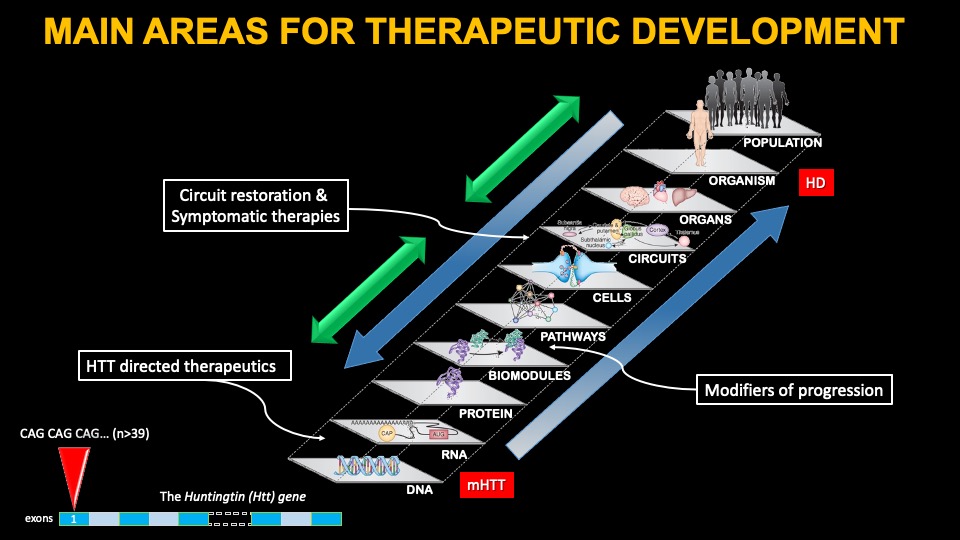
As duas áreas finais envolvem mecanismos de direcionamento ou genes que desempenham um papel causal na progressão da DH; o primeiro é o próprio gene HTT; a segunda, o direcionamento de outros genes identificados em pacientes humanos que progridem em taxas diferentes. Embora esta última área só agora esteja emergindo como uma nova estratégia terapêutica, ela está fazendo rápidos avanços em direção à clínica.
Abaixo você pode encontrar uma tabela abrangente mostrando todos os ensaios clínicos atuais em andamento ou recrutamento para o tratamento da doença de Huntington, com base em Clinicaltrials.gov
Você pode ver a tabela completa em PDF clicando nela.

Terapias genéticas direcionadas ao HTT
De longe, a principal área de investimento a desenvolver eficaz tratamentos para a DH tem como alvo a expressão do gene Huntingtin. Ampla pesquisa ao longo de muitos anos em modelos animais de DH apóia o direcionamento da expressão do HTT como uma terapia modificadora da doença. Sabemos com certeza em modelos de camundongos que, se alguém puder diminuir a expressão de HTT mutante o suficiente, os sintomas podem ser minimizados ou totalmente revertidos. No entanto, os modelos de DH em camundongos carecem de características significativas da doença – o mais importante, a morte de neurônios cerebrais e um ambiente inflamatório. Além disso, pessoas com DH nos estágios sintomáticos apresentam perda muito extensa de neurônios em várias áreas corticais e da maioria dos núcleos dos gânglios da base, as regiões mais afetadas na DH. Portanto, devemos ser cautelosos ao assumir que o cérebro humano com DH responderá de forma semelhante à diminuição da expressão de HTT, dadas essas diferenças muito significativas.
A maioria das terapias atualmente em desenvolvimento (ver Tabela acima) tem como alvo ambas as cópias do gene HTT, tanto a versão mutante quanto a cópia normal (ou 'tipo selvagem'). Há uma preocupação significativa de que a redução da cópia normal possa levar a efeitos indesejáveis após silenciamento prolongado. Sabemos também, por meio de estudos com camundongos, que a perda completa de ambas as cópias do HTT é letal durante o desenvolvimento embrionário. No adulto, os resultados são mais mistos e a perda significativa (mas não completa) do HTT pode ser melhor tolerada. Em humanos, sabemos que alguns indivíduos foram encontrados com apenas uma cópia do gene HTT e parecem estar bem. no entanto, indivíduos com mutações em ambas as cópias do gene HTT e que apresentam expressão muito baixa de HTT desenvolvem anormalidades cerebrais durante a infância. Portanto, diminuir demais o HTT provavelmente também é deletério em humanos.
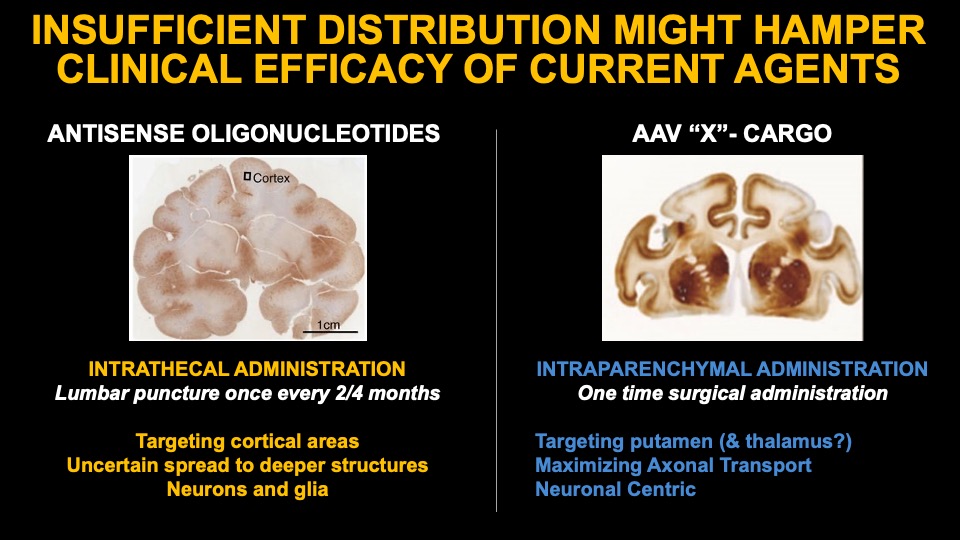
Este é um problema potencial isso pode ter prejudicado o programa Ionis/Roche, que na Fase 3 dos ensaios clínicos foi interrompido devido a eventos adversos. Neste ensaio, que gerou muitas expectativas e esperanças para uma primeira terapia modificadora da doença na DH, os indivíduos expostos à dose mais elevada de Tominersen (um oligonucleótido anti-sentido ou ASO) exibiram um agravamento na maioria das medidas clínicas da DH, aumento do tamanho do cérebro ventrículos e elevação das medidas de toxicidade e inflamação conforme julgado na análise de coleta de CSF, levando ao término prematuro do estudo.
Na mesma época, em março de 2021, dois outros programas de ASO em desenvolvimento pela Wave Therapeutics também foram interrompidos, desta vez na Fase 1/2, devido à falta de efeitos farmacológicos da expressão de mHTT no LCR de pacientes dosados. Em comparação com o teste da Roche com Tominersen, os Wave ASOs falharam em reduzir o mHTT o suficiente no CSF, levando a empresa a interromper seu desenvolvimento.
No entanto, é difícil saber por que Tominersen levou aos infelizes eventos descobertos no estudo Ionis/Roche – isso pode ser explicado por uma perda da função normal do HTT devido ao rebaixamento excessivo em algumas partes da medula espinhal ou do córtex cerebral (o áreas mais visadas por essas infusões da medula espinhal); mas também pode ser uma consequência do agente empregado: os oligonucleotídeos antisense podem ser pró-inflamatórios e seu acúmulo após dosagens repetidas também pode explicar alguns dos problemas encontrados. Não sabemos neste momento.
O próximo programa mais avançado na clínica no momento, o AMT130 da Uniqure, é um agente de entrega viral, um microRNA (mRNA) também visando ambas as cópias do gene HTT, atualmente em estudos de Fase 2. Esperamos uma divulgação inicial dos dados no final de 2022, embora o estudo tenha duração de cinco anos. Esta terapia AAV (vírus adeno-associado) é invasiva, requer neurocirurgia e é administrada diretamente no caudado e no putâmen, as duas estruturas dos gânglios da base do cérebro mais afetadas na DH. Estudos pré-clínicos em roedores, primatas não humanos e porcos mostraram que a terapia é bem tolerada por até 1 ano. Devido às propriedades do sorotipo AAV empregado (AAV-5), o agente terapêutico distribui-se amplamente por todo o cérebro, sendo transmitido através das fibras cerebrais que passam e inervam o corpo estriado. Veremos se esta terapia é bem tolerada a longo prazo.
Outros esforços de terapia genética também empregam AAVs, embora cada empresa que busca esses agentes tenha escolhido tipos virais diferentes; no caso do Voyager, com IND aberto e com previsão de iniciar estudos clínicos em HD em 2021, eles escolheram o AAV-1. O agente distribuído por meio desse vírus também tem como alvo as duas cópias do gene HTT e, portanto, será importante comparar seus efeitos com os do programa Uniqure.
Finalmente, a última empresa a provavelmente iniciar testes de terapia gênica em HD com um agente seletivo mHTT é a Takeda, que está desenvolvendo um agente repressor de dedo de zinco (ZFP) que diminui seletivamente de forma muito significativa a expressão de mHTT sem afetar a expressão normal de HTT, também sendo entregues por AAV-9. Há muita expectativa sobre esta terapia, que foi inicialmente desenvolvida pela Sangamo Therapeutics, pois é a única terapia alelo-seletiva em desenvolvimento. Este programa, se for bem-sucedido, levará a um IND no início de 2022.
Insira medicamentos orais de moléculas pequenas que reduzem o HTT
Os últimos agentes terapêuticos a surgir são uma nova classe de agentes orais de pequenas moléculas que visam a expressão do HTT em todo o corpo. Esses agentes foram inicialmente identificados em triagens fenotípicas visando os níveis de expressão do gene do neurônio motor espinhal (SMN)-2, como um tratamento para a atrofia muscular espinhal (AME).
Branaplam (também chamado LIM070) é um medicamento da Novartis que atua para aumentar os níveis de proteína SMN. Atualmente, o branaplam está na fase 2 para o tratamento da SMA. A análise de seletividade mostrou que o branaplam pode diminuir a expressão de ambas as cópias da proteína HTT via modulação de splicing do mRNA do HTT. O mecanismo de ação do branaplam parece levar ao decaimento do mRNA do HTT e à diminuição dos níveis de proteína. Recentemente, a Novartis recebeu a designação de medicamento órfão da FDA dos EUA para branaplam em HD, e um estudo de fase 2b está planejado para 2021.
Outra empresa que busca moduladores de splicing para reduzir o HTT é a terapêutica PTC. A empresa está realizando um teste de Fase 1 com seu medicamento PTC518 em voluntários saudáveis, esperando resultados em 2021, com um possível teste de Fase 2 no final do ano.
A entrada desses agentes na DH possibilita, pela primeira vez, testar drogas penetrantes orais e cerebrais para diminuir a expressão do HTT em todo o corpo, o que contorna os problemas de distribuição encontrados nos agentes de terapia gênica. No entanto, resta saber se a redução crônica e sistêmica de ambos os alelos do HTT gene será bem tolerado em pacientes adultos.
O impacto da genética humana no desenvolvimento de novas terapêuticas para a DH
Nos últimos dois anos, foram identificados novos alvos que parecem estar implicados na lentidão ou rapidez com que uma pessoa com a mutação avança para um estágio clínico. A idade de início (AOO) na DH é definida como a data em que um neurologista clínico diagnostica um indivíduo com sintomas motores de DH. Este 'marco' na progressão da DH foi escolhido como um momento importante no avanço da doença para avaliar se as influências genéticas podem afetar a taxa de progressão. Foi bem conhecido durante muitos anos que alguns indivíduos podem desenvolver sintomas motores da DH muito antes ou muito mais tarde do que a 'média' dos indivíduos positivos para a DH com uma mutação com o mesmo comprimento de repetições CAG no gene HTT. Os grupos de pesquisa conseguiram identificar cerca de uma dúzia de genes associados a essa mudança na taxa média de progressão. Este trabalho, em um grande estudo GWAS (genome-wide Association Study), levou à identificação de genes implicados no reparo do DNA e na expansão das repetições CAG em células somáticas (todas as outras células além das células reprodutivas do corpo). A compreensão mecanicista de como potencialmente esses genes modificam a progressão da DH antes dos sintomas clínicos levou algumas empresas a tentar desenvolver terapias direcionadas a esse mecanismo. O programa mais avançado até agora tem como alvo a expressão do gene Msh3, que demonstrou modular a instabilidade somática da repetição HTT CAG. A Triplet é uma empresa que desenvolve ASOs visando o Msh3 e planeja registrar um IND em 2021.