
Sandrine Humbert do INSERM, Grenoble Institute for Neurosciences, falou sobre o papel do tipo selvagem HTT no desenvolvimento do cérebro. Sandrine postula que um componente significativo da patogênese da DH envolve a perda da função da(s) função(ões) HTT normal(is), que envolvem, entre outras funções, a montagem de complexos moleculares, incluindo aqueles transportados por microtúbulos, e no contexto da ciliogênese. Ela também especula que um evento patológico primário é a perda de suporte aferente cortical para os neurônios de produção espinhosa do estriado, em parte por uma perda de suporte neurotrófico impulsionado pela sinalização de BDNF/TrkB. Sandrine revisou o trabalho anterior que sustentava essas hipóteses, que dão credibilidade ao conceito de que as funções de desenvolvimento do HTT são afetadas na DH. Um dos principais efeitos da perda do HTT normal durante o desenvolvimento são as alterações na progressão do ciclo celular durante a neurogênese (menos neurônios sendo gerados) e mudanças na polaridade neuronal, que se refletem em muitos contextos diferentes durante o desenvolvimento dos neurônios corticais, inclusive na morfologia dendrítica em neurônios pós-mitóticos.
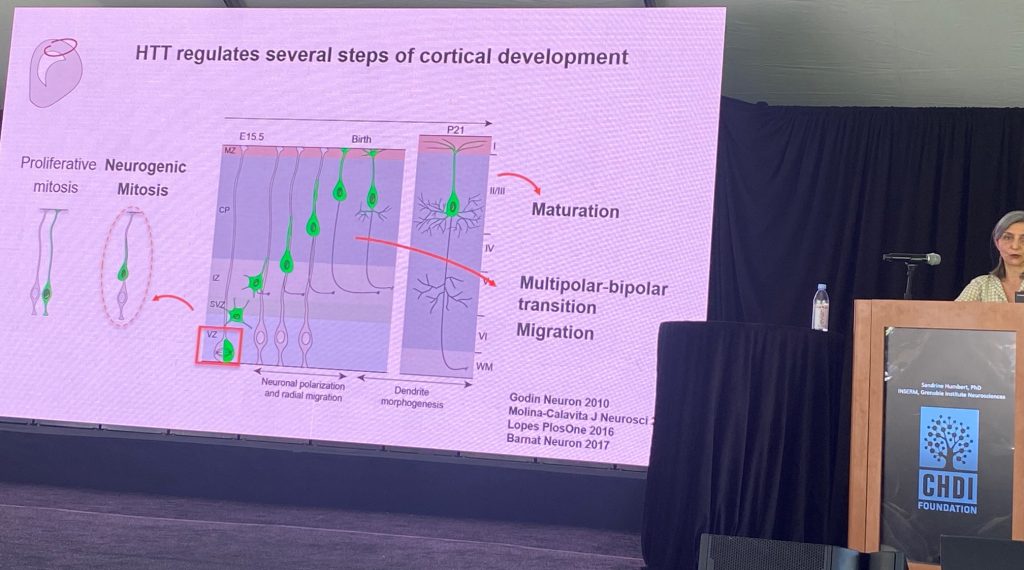
Alguns dos déficits observados nos nocautes de HTT de roedores parecem ser encontrados também em análises histológicas realizadas em tecidos fetais humanos, embora o tamanho da amostra seja pequeno. Parece haver evidências convergentes de uma mudança na localização do HTT na DH e uma mudança na polaridade celular no início do desenvolvimento do córtex, bem como em déficits no número e na morfologia dos cílios nas células progenitoras corticais. Outras observações aparentemente compartilhadas entre modelos de roedores e indivíduos com DH incluem agenesia parcial do corpo caloso (ou seja, que parte da bem conhecida perda de fibras calosas na DH se deve a um problema de crescimento axonal durante o desenvolvimento). Sandrine então descreveu o papel da proteína NUMA1 nos déficits do cone de crescimento axonal em neurônios derivados de modelos de camundongos knock-in. A superexpressão de NUMA1 em camundongos Q111 pode restaurar os déficits de desenvolvimento caloso neste modelo. Eles também mostraram que a epotilonaB, um produto natural que interfere na polimerização da tubulina e também leva a melhorias na estrutura calosa neste modelo.
RAFAELLE LENNACO
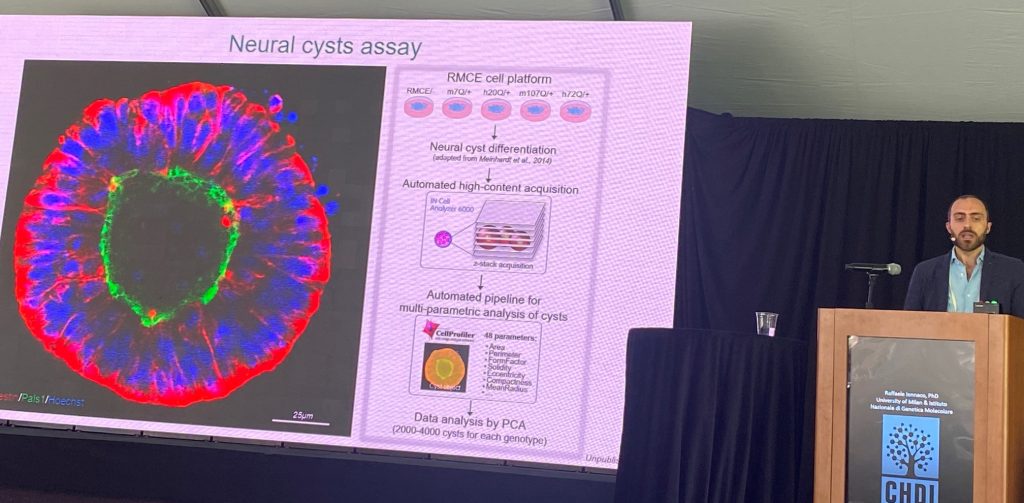
Rafaelle Iennaco (pós-doutorado do grupo da Dra. Elena Cattaneo), University of Milano falou sobre o papel de resíduos específicos dentro do exon-1 do gene HTT na modulação de sua função. Rafaelle projetou células-tronco de camundongos expressando várias deleções e mutações pontuais no gene HTT e sua relação com sua função de desenvolvimento. Na evolução, o número de repetições CAG começa a aumentar em aves, répteis, peixes e anfíbios (4Qs), enquanto nos mamíferos o intervalo é entre 5-35 repetições CAG. O N17 é muito conservado, mas o domínio poli-prolina (o 'PRD') também é altamente divergente ao longo da evolução, que só aparece em mamíferos. No entanto, o papel dessas 3 regiões e a importância de resíduos específicos não foram suficientemente aprofundados. Portanto, eles projetaram um grande conjunto de células MES que expressam vários comprimentos do trato polyCAG (Q = 0, 2, 4, 7, 17 etc.) e mostraram afetar um defeito de desenvolvimento precoce observado na cultura. eles também identificaram um conjunto de genes cuja expressão se correlacionava com o número de CAGs na faixa normal, revelando genes implicados no desenvolvimento do sistema nervoso.
A próxima etapa da apresentação focou na compreensão do papel patogênico do mutante humano HTT (mais patogênico que o éxon 1 do camundongo). Eles compararam os mesmos intervalos patogênicos de repetições CAG no contexto humano versus o contexto do camundongo, no contexto da morfologia de 'cistos' neuronais e em morfologias dendríticas em cultura, apoiando que as sequências humanas são importantes para desencadear esses déficits. Uma vez que as principais alterações entre o exon-1 do HTT de camundongo e humano residem na região de poliprolina, um conjunto de células-tronco knock-in foi gerado onde as regiões de polipro foram trocadas entre humano e camundongo. a região de poliprolina humana é importante para a patogênese nesses dois sistemas. A região humana piora e a região do camundongo melhora os fenótipos descritos.
Dr. BALJIT CAQUI
Dr. Baljit Khaki da UCLA descreveu seu trabalho sobre o impacto patogênico do mHTT em modelos de camundongos R6/2 e Q175, em termos de déficits específicos de células, comparando o papel patogênico do mHTT em astrócitos versus neurônios no corpo estriado (em nível fisiológico, molecular e comportamental) . Mal é um líder mundial em fisiologia de astrócitos e seu papel nas funções normais do cérebro e na doença. Neste seminário, Bal mostrou que muitos desses déficits podem ser evitados/restaurados depois que as células estriadas são expostas a um repressor de proteína dedo de zinco (ZFP) específico para mHTT que inibe a expressão de mHTT especificamente sem afetar a expressão normal de HTT. A novidade de seu trabalho recente é o fato de sua equipe expressar o mHTT ZFP apenas em neurônios ou apenas em astrócitos, e ser capaz de analisar o impacto da supressão do mHTT em termos de mecanismos autônomos de células versus não autônomos celulares - apontando, portanto, para o tipo de célula mais importante na condução de mecanismos patogênicos. Ou seja, diminuir mHTT em astrócitos e estudar efeitos em astrócitos e em neurônios (que ainda expressam mHTT), e vice-versa. Seus resultados suportam de forma esmagadora que, em modelos de HD de camundongos, a expressão neuronal de mHTT é o evento mais significativo na condução da progressão da doença, e que a maioria das alterações de astrócitos são adaptativas aos déficits desencadeados em neurônios estriatais por mHTT.

Bal também descreve o fato de que GPCRs astrocíticos que sinalizam via acoplamento Gi podem restaurar parcialmente os sintomas da doença em vários modelos de HD, demonstrando que esta é uma estratégia terapêutica potencialmente valiosa. Ele expandiu esses esforços anteriores, publicados em 2021, para estudar o papel dos efeitos de supressão do mHTT em neurônios versus astrócitos para entender os mecanismos pelos quais o mHTT contribui para a doença e os tipos de células mais importantes para a doença. As ZFPs foram expressas em vírus AAV e usadas extensivamente na Vivo em vários estágios de desenvolvimento da doença. Uma mensagem importante é que as alterações nos astrócitos relacionadas ao metabolismo do colesterol são confirmadas e que essa via pode ser resgatada de forma autônoma pelas células quando o mHTT é suprimido nos astrócitos. O laboratório de Bal também usou técnicas eletrofisiológicas para mostrar a restauração do disparo anormal e das propriedades celulares após a redução do mHTT neuronal, mas não astrocítica.

Um conjunto final de experimentos foi reduzir o mHTT em todo o cérebro usando o AAV PHPeb para direcionar a expressão do ZFP. Este tratamento mostrou melhorar as medidas da doença em camundongos R6.2, um modelo muito agressivo de HD. Esses estudos também mostraram que a expressão neuronal de ZFP, mas não astrocítica. expressão resgata os déficits comportamentais por múltiplos pontos finais (aninhamento, campo aberto, aperto, etc.).


