De uma mutação genética a uma doença letal
A DH é causada por uma única mutação em uma região do gene HTT, localizada no início do gene (no éxon 1), que contém uma sequência repetida composta por repetições CAG; na faixa normal, as pessoas têm em média entre 17 e 22 repetições; em indivíduos em Huntington, uma expansão superior a 39 leva inevitavelmente à doença. Quanto maior o número de repetições, normalmente, em média, mais cedo os pacientes começam a apresentar sintomas visíveis da doença.
Isto significa que todas as pessoas com DH têm o mesmo tipo de mutação em apenas um gene, tornando a DH uma doença incomum. A maioria das outras doenças neurodegenerativas comuns tem origens muito diferentes e apenas entre 2-5% de todos os casos de Alzheimer, Parkinson ou ELA têm uma base genética.
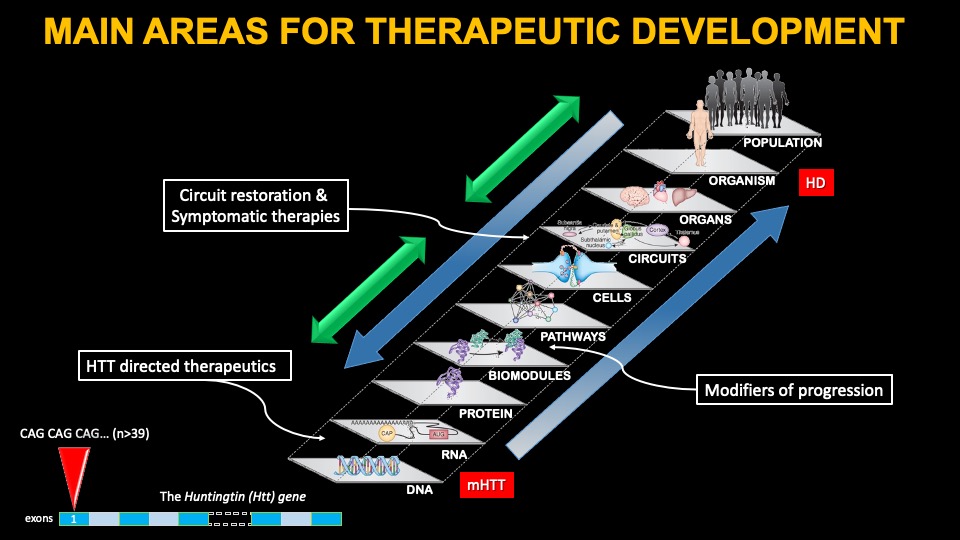
Muita investigação tem sido dedicada a descobrir como a mutação no gene HTT leva à DH, e muitos mecanismos celulares potenciais foram identificados. O impacto da mutação tem consequências devastadoras para muitas células cerebrais, e a teoria diz que, se conseguirmos compreender como isto acontece, poderemos ser capazes de desenvolver terapias para tratar a doença. No entanto, estes esforços têm sido em grande parte mal sucedidos até agora, incluindo resultados decepcionantes com abordagens de estimulação cerebral profunda. Algumas terapias sintomáticas, visando um ou outro mecanismo considerado errado em humanos afetados pela DH, estão em desenvolvimento, e abaixo cobrimos duas das terapias mais avançadas, Pridopidina e Valbenazina.
Outra abordagem para tratar a DH é visar sintomas específicos da doença que se pensa surgirem de alterações em circuitos cerebrais específicos. O HTT mutante leva à perda de células e fibras cerebrais em circuitos específicos do cérebro, o que explica em parte muitos dos sintomas da doença. Portanto, fortalecer ou restaurar esses circuitos pode oferecer algum benefício. No caso da DH, muitos circuitos são afetados, mas os principais envolvem a conexão entre várias áreas corticais e os gânglios da base, embora outras alterações sejam conhecidas no cerebelo e no hipotálamo.
Esta é a abordagem “tradicional” adotada para terapias sintomáticas em todas as doenças degenerativas e que tem sido eficaz no controle de alguns sintomas. Por exemplo, levou ao desenvolvimento da tetrabenazina para o controle da coreia e ao uso de medicamentos antipsicóticos direcionados ao sistema dopaminérgico para irritabilidade, por exemplo. No entanto, neste momento, faltam terapias que visem os sintomas mais debilitantes da DH – alterações cognitivas e apatia.
Notícias sobre tratamentos sintomáticos
Pridopidina, formalmente conhecido como ACR16, está sendo perseguido pela empresa Prilênia. ACR16 foi desenvolvido anteriormente pela Teva. Num estudo de Fase III recentemente concluído (PIVOT-HD), a Pridopidina não conseguiu atingir o objetivo primário do ensaio, mas mostrou sinais de melhoria de alguns sintomas nos domínios cognitivo e motor. apenas no subconjunto de pacientes em HD que não tomam medicamentos antidopaminérgicos, como tetrabenazina ou antipsicóticos. The primary endpoint was change from baseline to week 65 in TFC (the total functional capacity test). The key secondary endpoint was change from baseline in the composite Unified Huntington Disease Rating Scale (cUHDRS), and additional endpoints included quantitative motor (Q-Motor), the cognitive test Stroop Word Reading (SWR), and Quality of Life (HD-QoL). The Phase III study enrolled 499 individuals and was conducted in the U.S., Canada, Austria, Czech Republic, France, Germany, Italy, the Netherlands, Poland, Spain and the United Kingdom.
Prilenia is now appealing to regulatory authorities to enable a registration path for Pridopidine without having to conduct new clinical trials. Currently, they are awaiting feedback from the European Regulatory Agency (EMA) to see if their application for Market Authorization (MAA) is approved, which would enable the company to market the product in Europe.
A pridopidina foi investigada como tratamento para a DH em três ensaios clínicos randomizados, duplo-cegos e controlados por placebo: HART, MermaiHD e PRIDE-HD. Os estudos iniciais na DH centraram-se nos efeitos da pridopidina nos parâmetros motores, sob a hipótese de que a pridopidina tinha um efeito no controlo dopaminérgico do movimento. De fato, um pequeno efeito nos endpoints motores foi observado nos estudos HART e MermaiHD, embora as pessoas também tenham visto um pequeno efeito na escala de capacidade funcional total (TFC) e estenderam o estudo de fase 2 em uma modificação aberta para 52 semanas.
No estudo PRIDE-HD, a dosagem de pridopidina demonstrou um efeito pequeno mas benéfico no TFC e este efeito pareceu ser mais pronunciado para os participantes iniciais da DH, o que levou os investigadores a alargar estas observações num estudo de Fase 3, PROOF-HD, actualmente a incluir e usando a pontuação UHDRS-TFC como o endpoint primário.
Valbenazina é um medicamento recentemente aprovado pela FDA (nome comercial INGREZZA) após resultados positivos em coreia em HD no ensaio de Fase III (KINECT-HD) desenvolvido pela empresa Biociências Neurócrinas. You can see the PDF of the news release AQUI.
Os principais resultados dos ensaios clínicos do KINECT-HD incluem:
- A three-times greater improvement in chorea compared to placebo, from the start to the end of the 12-week clinical study. A reduction in chorea severity by about 40 percent from baseline to maintenance, and nearly half of patients saw a more than 40 percent reduction in chorea severity by Week 12.1
- Cinquenta e três por cento dos doentes e 43 por cento dos profissionais de saúde relataram que os sintomas globais da coreia da DH estavam “muito melhorados” ou “muito melhorados”
A valbenazina funciona de forma semelhante à tetrabenazina, mas oferece algumas vantagens, nomeadamente administração oral uma vez ao dia e menor perfil de eventos adversos. Resta saber como os pacientes se comportam quando tomam Valbenazina por longos períodos de tempo.
Terapias genéticas direcionadas ao HTT
By far the major area of investment to develop effective treatments for HD is targeting the expression of the Huntingtin gene. Ample research over many years in animal models of HD support the targeting of HTT expression as a disease modifying therapy. We know with certainty in mouse models that if one can decrease mutant HTT expression enough, manifestations can be minimized or altogether reversed. However, mouse models of HD lack significant features of the disease – most importantly, the death of brain neurons and an inflammatory environment. In addition, people with HD in the symptomatic stages have very extensive loss of neurons in several cortical areas and most nuclei of the basal ganglia, the regions most affected in HD. Therefore, we must be cautious assuming that the human HD brain will respond similarly to lowering HTT expression, given these very significant differences.
Most therapies currently under development target both copies of the HTT gene, both the mutated version and the normal (or ‘non-expanded’) copy. There is significant concern that lowering of the normal copy might lead to untoward effects after prolonged silencing. We know also from mouse studies that a complete loss of both copies of HTT is lethal during embryonic development. In the adult, the results are more mixed and significant loss (but not complete) of HTT can be better tolerated. In humans, we know that a few individuals have been found with only one copy of the HTT gene, and they do not exhibit the typical clinical manifestations of HD. However, individuals with mutations in both copies of the HTT gene and that show very low expression of HTT develop brain abnormalities during childhood. Therefore, lowering HTT too much is likely deleterious in humans as well.
Terapêutica ASO em desenvolvimento clínico
This is a potential issue that might have derailed the Ionis/Roche Tominersen Programa ASO, que nos ensaios clínicos de Fase 3 foi interrompido devido a eventos adversos. Neste ensaio, que gerou muitas expectativas e esperança para uma primeira terapia modificadora da doença na DH, os indivíduos expostos à dose mais elevada de Tominersen (um oligonucleótido anti-sentido ou ASO) exibiram um agravamento na maioria das medidas clínicas da DH, aumento do tamanho do cérebro ventrículos e elevação das medidas de toxicidade e inflamação conforme julgado na análise da coleta de LCR, levando ao encerramento prematuro do estudo. A partir de 2024, a Roche está a recrutar para um novo estudo de Fase II com Tominersen com base numa análise post-hoc dos dados recolhidos no estudo de Fase III, que indicou que a DH afectava indivíduos com pontuações baixas de PAC (uma medida da progressão da doença baseada em idade e duração das repetições CAG) e os jovens toleraram mais a droga. Você pode ler o raciocínio da Roche neste Artigo NEJM. Além de recrutar pacientes mais jovens e menos afetados, o estudo será realizado com uma dose mais baixa de tominersen, o que pode impactar a distribuição do medicamento em áreas cerebrais mais profundas afetadas na DH.
Mais ou menos na mesma época do fracasso de Tominersen, em março de 2021, dois outros programas ASO sendo desenvolvidos por Ciências da Vida das Ondas, também foram interrompidos, desta vez na Fase 1/2, devido à falta de efeitos farmacológicos da expressão do mHTT no LCR dos pacientes dosados. Em comparação com o ensaio da Roche com Tominersen, os Wave ASOs não conseguiram reduzir suficientemente o mHTT no LCR, levando a empresa a interromper o seu desenvolvimento. No entanto, a empresa perseverou e desenvolveu um terceiro ASO visando um polimorfismo de nucleótido único (SNP) encontrado num subconjunto de doentes de Huntington de ascendência europeia, denominado WVE-003. Este ASO, ao contrário do Roche ASO Tominersen, é seletivo para a cópia mutante do gene HTT. Na semana passada, a Wave anunciou resultados positivos de seu WVE-003 molécula em um estudo de Fase 1/2 (ensaio SELECT-HD), demonstrando segurança e uma redução significativa no mHTT no LCR do paciente (redução de 45%). Além disso, parece haver estabilização do volume caudado e do escore funcional total. Você pode ler o comunicado de imprensa AQUI.
Esta é realmente uma notícia muito animadora, pois é o primeiro caso em que uma terapia seletiva de mHTT demonstrou um efeito na redução do mHTT em pacientes em HD. O ensaio também mostrou que a terapia com WVE-003 foi bem tolerada e não relatou aumento de volumes ventriculares, como aqueles comumente observados com Tominersen. Estamos aguardando a divulgação pública dos resultados do ensaio, incluindo dados clínicos e de segurança sobre os principais desfechos funcionais e motores.
É difícil saber porque é que Tominersen levou aos acontecimentos infelizes descobertos no ensaio Ionis/Roche – isto poderia ser explicado por uma perda da função normal do HTT devido ao abaixamento excessivo em algumas partes da medula espinal ou do córtex cerebral (as áreas mais alvo destas infusões da medula espinhal); mas também pode ser uma consequência do agente utilizado: os oligonucleótidos antisense podem ser pró-inflamatórios, e a sua acumulação após doses repetidas também pode explicar alguns dos problemas encontrados. Não sabemos neste momento. Se a abordagem seletiva do mHTT funcionar melhor e for melhor tolerada, isso poderá apoiar a suposição de que uma supressão global do HTT normal não é bem tolerada nas pessoas, e isso afetará muitos outros programas que visam reduzir as cópias mutantes e normais do gene HTT.
Programas de terapia genética em desenvolvimento clínico
O próximo programa mais avançado da clínica no momento, Exclusivode AMT130, é um agente distribuído por vírus, um microRNA (miRNA) que também tem como alvo ambas as cópias do gene HTT, atualmente em estudos de Fase 2. Você pode ver um bom vídeo descrevendo essa abordagem AQUI.
Esperamos a divulgação dos dados do ensaio no final de 2024, embora o ensaio tenha uma duração de cinco anos e tenha começado em 2021. Um total de 26 pacientes foram inscritos nos EUA e na Europa. Esta terapia com AAV (vírus adeno-associado) é invasiva, requer neurocirurgia e é administrada diretamente no caudado e no putâmen, as duas estruturas dos gânglios da base do cérebro mais afetadas na DH. Estudos pré-clínicos em roedores, primatas não humanos e porcos demonstraram que a terapia é bem tolerada até 1 ano. Devido às propriedades do serótipo AAV utilizado (AAV-5), o agente terapêutico distribui-se amplamente por todo o cérebro, sendo transmitido através das fibras cerebrais que passam e inervam o corpo estriado. Veremos se esta terapia é bem tolerada a longo prazo, mas os dados apresentados pela UniQure não mostraram nenhuma evidência de efeitos deletérios até o momento nas duas doses administradas (com várias quantidades do produto AAV-AMT130). Isto não deve ser dado como certo, sendo que este é o primeiro ensaio clínico de terapia genética com AAV na história da investigação na DH. O ambiente cerebral em degeneração é um ambiente complicado para terapias invasivas como esta devido a um sistema imunológico hiperativo desencadeado pela morte de células cerebrais; o fato de os pacientes tolerarem bem a cirurgia e a terapia é uma conquista por si só!
Recently, UniQure obtained from the FDA the Regenerative Medicine Advanced Therapy (RMAT) designation, laying the path for a Phase III trial in HD (see the press release AQUI). Como os estudos clínicos ainda estão em andamento (incluindo um braço onde os indivíduos recebem terapia com medicamentos imunossupressores), precisamos esperar até o final de 2024 para ver todo o pacote de dados deste importante ensaio.
Outros esforços de terapia genética também empregam AAVs, embora cada empresa que busca esses agentes tenha escolhido diferentes tipos virais; no caso de Viajante, eles escolheram AAV-1. O agente entregue através deste vírus também tem como alvo ambas as cópias do gene HTT e, portanto, será importante comparar os seus efeitos com os do programa Uniqure. Esperamos ver este programa na clínica em breve.
Finalmente, a última empresa que esperávamos iniciar ensaios de terapia genética na DH com um agente selectivo para mHTT era a Takeda, que estava a desenvolver um agente repressor de dedo de zinco (ZFP) que diminui selectivamente de forma muito significativa a expressão de mHTT sem afectar a expressão normal de HTT. , também sendo entregue pela AAV-9. Havia muita expectativa em relação a essa terapia, que foi inicialmente desenvolvida pela Sangamo Therapeutics, por ser a única terapia seletiva de alelos para uma ampla população em desenvolvimento. Infelizmente, Takeda anunciou a descontinuação de seus programas de terapia genética, incluindo o programa ZFP para HD.

Medicamentos orais de moléculas pequenas que reduzem o HTT
Os últimos agentes terapêuticos a surgir são uma nova classe de agentes orais de pequenas moléculas que visam a expressão do HTT em todo o corpo. Esses agentes foram inicialmente identificados em triagens fenotípicas visando os níveis de expressão do gene do neurônio motor espinhal (SMN)-2, como um tratamento para a atrofia muscular espinhal (AME).
Branaplam (também chamado de LIM070) é um medicamento da Novartis que atua aumentando os níveis da proteína SMN-2. O Branaplam foi avaliado na fase 2 para o tratamento da DH (o estudo VIBRANT-HD). A análise de seletividade mostrou que o Branaplam pode diminuir a expressão de ambas as cópias da proteína HTT através da modulação de splicing do mRNA do HTT. O mecanismo de ação do Branaplam parece ser levado ao decaimento do mRNA do HTT e à diminuição dos níveis de proteína. Em 2021, a Novartis recebeu a designação de medicamento órfão da FDA dos EUA para Branaplam em HD, mas o estudo de Fase 2b foi interrompido devido a eventos adversos (foi observada neuropatia periférica em alguns pacientes) e o programa foi descontinuado.
Outra empresa que busca moduladores de emenda para reduzir o HTT é Terapêutica PTC. A empresa está conduzindo um ensaio de Fase 2a (estudo PIVOT-HD) com seu medicamento PTC-518 em indivíduos de Huntington, esperando que os resultados sejam anunciados no final de 2024. Ainda esta semana, a PTC Therapeutics anunciou num comunicado de imprensa (ver AQUI) que o PTC-518 foi bem tolerado e reduziu os níveis de HTT (ambos os alelos) na periferia (medindo o HTT nas células sanguíneas) e no LCR (na dose mais alta de 10 mg, eles relataram uma redução de 43% no mHTT). Este é o primeiro medicamento oral de molécula pequena com distribuição em todo o corpo que mostra uma redução do HTT em todo o corpo em um estudo clínico. O PTC também relata uma desaceleração dos sintomas motores em pacientes que tomam a dose mais elevada de PTC-518.
A entrada desses agentes na DH possibilita, pela primeira vez, testar drogas penetrantes orais e cerebrais para diminuir a expressão do HTT em todo o corpo, o que contorna os problemas de distribuição encontrados nos agentes de terapia gênica. No entanto, resta saber se a redução crônica e sistêmica de ambos os alelos do HTT serão bem tolerados em pacientes adultos e levarão a efeitos sustentados modificadores da doença.
O impacto da genética humana no novo desenvolvimento terapêutico
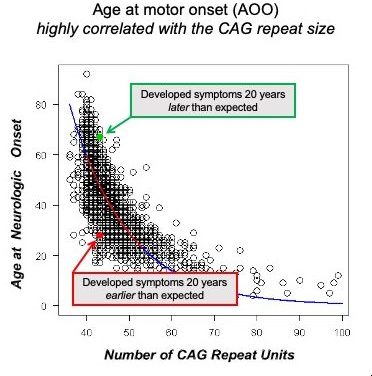
Nos últimos anos, foram identificados novos alvos que parecem estar implicados na lentidão ou rapidez com que uma pessoa com a mutação avança para um estágio clínico. A idade de início (AOO) na DH é definida como a data em que um neurologista clínico diagnostica um indivíduo com sintomas motores típicos da DH. Este “marco” na progressão da DH foi escolhido como um momento importante no avanço da doença para avaliar se as influências genéticas podem afectar a taxa de progressão. Era bem conhecido há muitos anos que alguns indivíduos podem desenvolver sintomas motores da DH muito mais cedo ou muito mais tarde do que a “média” dos indivíduos DH positivos com uma mutação com o mesmo comprimento de repetições CAG no gene HTT. Os grupos de pesquisa conseguiram identificar cerca de uma dúzia de genes associados a essa mudança na taxa média de progressão. Este trabalho, em um grande estudo GWAS (estudo de associação genômica ampla) (veja a última publicação AQUI), levou à identificação de genes implicados no reparo do DNA e na expansão das repetições CAG nas células somáticas (todas as outras células além das células reprodutivas do corpo). A compreensão mecanicista de como estes genes modificam potencialmente a progressão da DH antes dos sintomas clínicos levou algumas empresas a tentar desenvolver terapias direcionadas a este mecanismo. O programa mais avançado até agora tem como alvo a expressão do gene MSH3 ou a sua atividade, que demonstrou modular a instabilidade somática da repetição HTT CAG. Nenhum destes programas está ainda em desenvolvimento clínico, mas há progressos significativos no sentido de obter novas terapias direcionadas a este mecanismo na clínica.




