El Dr. José Lucas desde el Centro de Biología Molecular Severo Ochoa de Madrid habló acerca de este reciente trabajo en el papel de mHTT en la alteración de empalme y adenilación de arnm en HD. José comenzó describiendo su trabajo pionero en el tau empalme alteraciones en el HD del cerebro. También describió los cambios que empalma en TAF1 de proteínas, lo que conduce a la degeneración estriatal y discinesias en un raro desorden genético. Él postula que mHTT conduce a cambios en SRSF6 con múltiples efectos, uno para modular la Tau de empalme, y de la sec y, a un TAF1 la pérdida de la función. Debido a estas observaciones independientes, su laboratorio persigue un profundo análisis imparcial de proteínas de unión al ARN y global missplicing eventos que ocurren en HD. Él identificó 342 genes misspliced en humanos y roedores muestras, incluyendo varios genes que se sabe que conducen a la degeneración cuando está mutado. Él se centró, entonces, en la alteración de los factores de empalme que podría explicar los fenotipos observados, tales como TIA1, hNRPC, U2AF2, y RBFOX, el último siendo un factor clave que está regulado en HD. Ellos crearon un ratón transgénico que la sobre-expresión de RBFOX1 y cruzó a la R6/1 modelo de ratón, que conduce a una atenuación de la enfermedad. Él se refirió a la labor realizada en 2011 por el Dr. Nancy Bonini y se centró en un conjunto de genes que se sabe moderada ARN toxicidad en las moscas, incluyendo musclebound y la orb2 (CPEB4 es el mamífero ortólogo), un gen que regula la poliadenilación. CPEB proporciona también la regulación de la traducción en las sinapsis de una manera temporal. Jose, a continuación, mostró que CPEB1 y -4 son alterados en HD, y se determinó que el 20% de las transcripciones de los cambios en adenilación, incluyendo SLC19A3, un transportador de tiamina en el cerebro del endotelio. Deficiencias en SLC19A3 puede ser restaurado por la administración de biotina, presumiblemente a través de un efecto sobre SLC19A3 niveles. Por lo tanto, si su hipótesis es correcta, entonces los pacientes en HD debe tener menos de tiamina concentración en el LCR y en el parénquima cerebral. Un conjunto final de los estudios mostraron que la tiamina y la suplementación con biotina, la terapia puede restaurar los cambios de comportamiento en R6/2 y Q175 modelos. Se ha presentado una patente, y un pequeño estudio clínico ha sido planeado (llamado el HUNTIAM de estudio) para evaluar la seguridad de esta estrategia de suplemento y la dosis que trabajan en los ratones son muy altos y mostraron algunas de toxicidad en los estudios con roedores. Trabajo adicional para extender la disminución de los niveles de tiamina en el LCR es de bering explorado en un estudio longitudinal con el hospital de Sant Pau en Barcelona.
El Dr. MICHAEL VIOLACIÓN
El Dr. Michael Violación de la Universidad de Berkeley y el Instituto Médico Howard Hughes habló acerca de los mecanismos que degradan la HTT proteína a través de la proteasoma, un paso crítico en la regulación del nivel de expresión de la proteína, y un área importante para el desarrollo terapéutico. Retos clave para esta área ha sido la identificación de la ubiquitina-ligasa complejos que pueden modificar HTT y de destino para la degradación, y la lisina específico de los residuos de aminoácidos que conseguir modificado y que regulan su vida media. Él describe a continuación, el reciente obra inédita que ha llevado a identificar el E3 ligasa que trabaja para modificar HTT directamente y degradan. Su equipo se ha reconstituido ubiquitinación HTT in vitro, abriendo el camino para la identificación de la HTT sitios modificados, y de un sistema de detección para identificar molecular colas pequeñas moléculas que promueven la interacción entre HTT y la identificación de las ligasa E3.

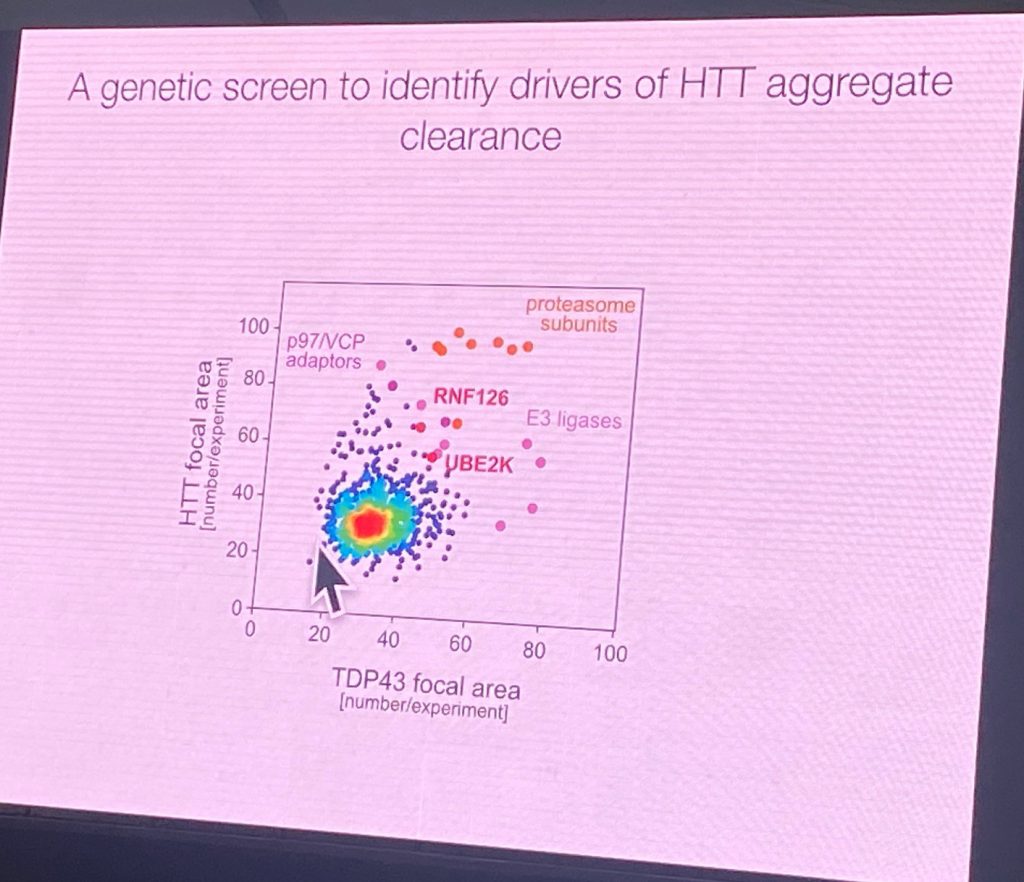
Él afirma que la degradación de las señales para HTT se encuentran en la región después de la primera CALOR de dominio de HTT. El uso de una fusión GFP con 508 AA N-terminal de la HTT y la realización de una pantalla en establemente transfectadas las células de neuroblastoma de identificar los factores que promueven la agregación de este fragmento (que normalmente no agregado). El uso de varias señales de estrés, agregados aparece en estas células, y después de la eliminación de los factores de estrés, los agregados ('focos') se borran. Extendida la exposición al estrés conduce a una CAG longitud dependiente de la agregación, incluso después de largos períodos de lavado. Esto permitió una ligasa E3 y efectores de la pantalla genómicas que condujo a la identificación de los complejos que pueden afectar este proceso, retrasar el despacho de la HTT, RNF126b y UBE2K. RNF126 se une directamente a la HTT y el p97/VCP disaggregase. También es altamente expresado en el cerebro. Además, la Violación del laboratorio demostraron que un ensayo de degradación puede ser desarrollado para mostrar dirigir la actividad de RNF126 complejos en HTT en las células. RNF126 KO aumento de la agregación de HTT, y la sobre-expresión puede degradar en un contexto celular. Además, una reconstituido ubiquitinación del sistema se logró con purificado RNF126 y HTT. Punto de mutantes que inactiva el RNF12 actividad catalítica de no tener tal actividad. Las enzimas E2 son UBE2K y UBE2D3. Él también mostró que este sistema también funciona con longitud completa HTT ubiquitinación, no sólo en el más corto reportero del sistema.
Su trabajo sugiere que existe un sistema para el control de la agregación de HTT y por lo tanto abierto el camino para la identificación de moléculas pequeñas que pueden orientar HTT para la degradación.