De una mutación genética a una enfermedad letal
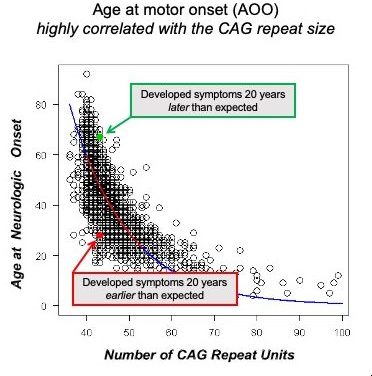
La EH es causada por una única mutación en una región del gen HTT, ubicada al comienzo del gen (en el exón 1), que contiene una secuencia repetida compuesta de repeticiones CAG; en el rango normal, las personas tienen entre 17 y 22 repeticiones en promedio; en personas con EH, una expansión superior a 39 conduce inevitablemente a la enfermedad. Cuanto mayor sea el número de repeticiones, por lo general, en promedio, antes los pacientes comenzarán a mostrar síntomas visibles de la enfermedad.
Esto significa que cada persona con EH tiene el mismo tipo de mutación en un solo gen, lo que hace que la EH sea un trastorno inusual. La mayoría de las otras enfermedades neurodegenerativas comunes tienen muchos orígenes diferentes, y sólo entre 2 y 5% de todos los casos de Alzheimer, Parkinson o ELA tienen una base genética.
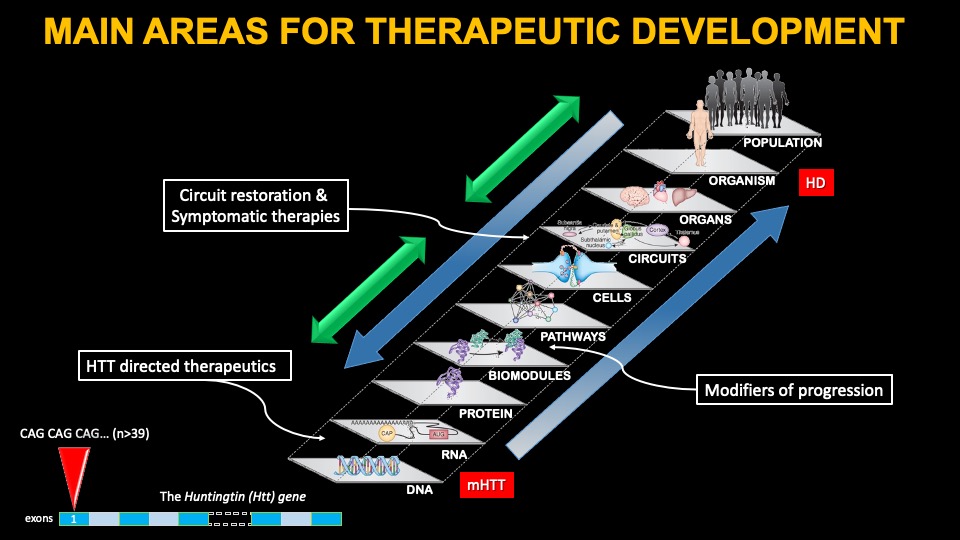
Se han dedicado muchas investigaciones a descubrir cómo la mutación en el gen HTT conduce a la EH y se han identificado muchos mecanismos celulares potenciales. El impacto de la mutación tiene consecuencias devastadoras para muchas células cerebrales y la teoría dice que si podemos entender cómo sucede esto, podríamos desarrollar terapias para tratar la enfermedad. Sin embargo, estos esfuerzos han sido en gran medida infructuosos hasta el momento, incluidos resultados decepcionantes con enfoques de estimulación cerebral profunda. Se están desarrollando algunas terapias sintomáticas, dirigidas a uno u otro mecanismo que se cree que funciona mal en humanos afectados con EH, y a continuación cubrimos dos de las terapias más avanzadas, Pridopidina y Valbenazina.
Otro enfoque para tratar la EH es enfocarse en síntomas específicos de la enfermedad que se cree que surgen de alteraciones en circuitos cerebrales específicos. La HTT mutante conduce a una pérdida de células y fibras cerebrales en circuitos específicos del cerebro, lo que explica parcialmente muchos de los síntomas de la enfermedad. Por lo tanto, fortalecer o restaurar esos circuitos podría ofrecer algún beneficio. En el caso de la EH, se ven afectados muchos circuitos, pero los principales involucran la conexión entre varias áreas corticales y los ganglios basales, aunque se conocen otros cambios en el cerebelo y el hipotálamo.
Este es el enfoque "tradicional" adoptado para las terapias sintomáticas en todos los trastornos degenerativos y que ha sido eficaz para controlar algunos síntomas. Por ejemplo, condujo al desarrollo de la tetrabenazina para el control de la corea y al uso de medicamentos antipsicóticos dirigidos al sistema dopaminérgico para la irritabilidad, por ejemplo. Sin embargo, por el momento, carecemos de terapias dirigidas a los síntomas más debilitantes de la EH: las alteraciones cognitivas y la apatía.
Novedades sobre tratamientos sintomáticos
pridopidina, formalmente conocido como ACR16, está siendo perseguido por la empresa Prilenia. ACR16 fue desarrollado anteriormente por Teva. En un estudio de fase III (PIVOT-HD) recientemente completado, la pridopidina no logró cumplir el criterio de valoración principal del ensayo, pero mostró signos de mejora de algunos síntomas en los dominios cognitivo y motor. sólo en el subconjunto de pacientes con EH que no toman medicamentos antidopaminérgicos como tetrabenazina o antipsicóticos. The primary endpoint was change from baseline to week 65 in TFC (the total functional capacity test). The key secondary endpoint was change from baseline in the composite Unified Huntington Disease Rating Scale (cUHDRS), and additional endpoints included quantitative motor (Q-Motor), the cognitive test Stroop Word Reading (SWR), and Quality of Life (HD-QoL). The Phase III study enrolled 499 individuals and was conducted in the U.S., Canada, Austria, Czech Republic, France, Germany, Italy, the Netherlands, Poland, Spain and the United Kingdom.
Prilenia is now appealing to regulatory authorities to enable a registration path for Pridopidine without having to conduct new clinical trials. Currently, they are awaiting feedback from the European Regulatory Agency (EMA) to see if their application for Market Authorization (MAA) is approved, which would enable the company to market the product in Europe.
La pridopidina se ha investigado como tratamiento para la EH en tres ensayos clínicos aleatorizados, doble ciego, controlados con placebo: HART, MermaiHD y PRIDE-HD. Los estudios iniciales en HD se centraron en los efectos de la pridopidina en los puntos finales motores, bajo la hipótesis de que la pridopidina tenía un efecto sobre el control dopaminérgico del movimiento. De hecho, se observó un pequeño efecto en los criterios de valoración motores en los estudios HART y MermaiHD, aunque las personas también observaron un pequeño efecto en la escala de capacidad funcional total (TFC) y ampliaron el estudio de fase 2 en una modificación abierta a 52 semanas.
En el estudio PRIDE-HD, la dosificación de pridopidina demostró un efecto pequeño pero beneficioso sobre el TFC y este efecto pareció ser más pronunciado para los primeros participantes de HD, lo que llevó a los investigadores a ampliar estas observaciones en un estudio de fase 3, PROOF-HD, que actualmente está reclutando y utilizando la puntuación UHDRS-TFC como criterio principal de valoración.
Valbenazina es un fármaco recientemente aprobado por la FDA (nombre comercial INGREZZA) tras resultados positivos sobre corea en EH en el ensayo de Fase III (KINECT-HD) desarrollado por la empresa Biociencias neurocrinas. You can see the PDF of the news release AQUÍ.
Los resultados clave de los ensayos clínicos de KINECT-HD incluyen:
- A three-times greater improvement in chorea compared to placebo, from the start to the end of the 12-week clinical study. A reduction in chorea severity by about 40 percent from baseline to maintenance, and nearly half of patients saw a more than 40 percent reduction in chorea severity by Week 12.1
- El cincuenta y tres por ciento de los pacientes y el 43 por ciento de los profesionales de la salud informaron que los síntomas generales de la corea de la EH habían "mejorado mucho" o "mucho mejorado".
La valbenazina funciona de manera similar a la tetrabenazina, pero ofrece algunas ventajas, como la administración oral una vez al día y un perfil de eventos adversos menor. Queda por ver cómo les va a los pacientes cuando toman valbenazina durante períodos prolongados.
Terapias génicas dirigidas a HTT
By far the major area of investment to develop effective treatments for HD is targeting the expression of the Huntingtin gene. Ample research over many years in animal models of HD support the targeting of HTT expression as a disease modifying therapy. We know with certainty in mouse models that if one can decrease mutant HTT expression enough, manifestations can be minimized or altogether reversed. However, mouse models of HD lack significant features of the disease – most importantly, the death of brain neurons and an inflammatory environment. In addition, people with HD in the symptomatic stages have very extensive loss of neurons in several cortical areas and most nuclei of the basal ganglia, the regions most affected in HD. Therefore, we must be cautious assuming that the human HD brain will respond similarly to lowering HTT expression, given these very significant differences.
Most therapies currently under development target both copies of the HTT gene, both the mutated version and the normal (or ‘non-expanded’) copy. There is significant concern that lowering of the normal copy might lead to untoward effects after prolonged silencing. We know also from mouse studies that a complete loss of both copies of HTT is lethal during embryonic development. In the adult, the results are more mixed and significant loss (but not complete) of HTT can be better tolerated. In humans, we know that a few individuals have been found with only one copy of the HTT gene, and they do not exhibit the typical clinical manifestations of HD. However, individuals with mutations in both copies of the HTT gene and that show very low expression of HTT develop brain abnormalities during childhood. Therefore, lowering HTT too much is likely deleterious in humans as well.
Terapéutica ASO en desarrollo clínico.
This is a potential issue that might have derailed the Ionis/Roche Tominersen Programa ASO, que en ensayos clínicos de Fase 3 se detuvo debido a eventos adversos. En este ensayo, que generó muchas expectativas y esperanzas para una primera terapia modificadora de la enfermedad en la EH, los sujetos expuestos a la dosis más alta de Tominersen (un oligonucleótido antisentido o ASO) mostraron un empeoramiento en la mayoría de las medidas clínicas de la EH, agrandamiento del cerebro. ventrículos y elevación de las medidas de toxicidad e inflamación según lo juzgado en el análisis de la recolección de LCR, lo que llevó a que el ensayo se terminara prematuramente. A partir de 2024, Roche está reclutando para un nuevo estudio de Fase II con Tominersen basado en un análisis post-hoc de los datos recopilados en el estudio de Fase III, que indicó que la EH afectaba a individuos con puntuaciones bajas de CAP (una medida de la progresión de la enfermedad basada en edad y duración de las repeticiones CAG) y los jóvenes toleraron más el fármaco. Puede leer el razonamiento de Roche en este Artículo del NEJM. Además de reclutar pacientes más jóvenes y menos afectados, el estudio se llevará a cabo con una dosis más baja de tominersen, lo que puede afectar la distribución del fármaco a áreas más profundas del cerebro afectadas en la EH.
Casi al mismo tiempo del fracaso de Tominersen, en marzo de 2021, otros dos programas ASO desarrollados por Ciencias de la vida de las olas, también se detuvieron, esta vez en la Fase 1/2, debido a la falta de efectos farmacológicos de la expresión de mHTT en el LCR de los pacientes que recibieron la dosis. En comparación con el ensayo de Roche con Tominersen, los Wave ASO no lograron reducir el mHTT lo suficiente en el LCR, lo que llevó a la empresa a detener su desarrollo. Sin embargo, la empresa perseveró y desarrolló un tercer ASO dirigido a un polimorfismo de un solo nucleótido (SNP) encontrado en un subconjunto de pacientes con EH de ascendencia europea, denominado WVE-003. Este ASO, a diferencia del ASO Tominersen de Roche, es selectivo para la copia mutante del gen HTT. La semana pasada, Wave anunció resultados positivos de su WVE-003 molécula en un estudio de fase 1/2 (ensayo SELECT-HD), que demuestra seguridad y una reducción significativa del mHTT en el LCR del paciente (disminución de 45%). Además, parece haber una estabilización del volumen caudado y de la puntuación funcional total. Puedes leer la nota de prensa. AQUÍ.
De hecho, esta es una noticia muy interesante, ya que es la primera vez que una terapia selectiva de mHTT ha demostrado un efecto en la reducción de mHTT en pacientes con EH. El ensayo también demostró que la terapia WVE-003 fue bien tolerada y no informó un aumento de los volúmenes ventriculares como los que se observan comúnmente con Tominersen. Estamos esperando la divulgación pública de los resultados del ensayo, incluidos los datos clínicos y de seguridad sobre criterios de valoración funcionales y motores clave.
Es difícil saber por qué Tominersen provocó los desafortunados acontecimientos descubiertos en el ensayo de Ionis/Roche; podría explicarse por una pérdida de la función HTT normal debido a una disminución excesiva en algunas partes de la médula espinal o de la corteza cerebral (las áreas más objetivo de estas infusiones de médula espinal); pero también podría ser una consecuencia del agente empleado: los oligonucleótidos antisentido pueden ser proinflamatorios y su acumulación después de dosis repetidas también podría explicar algunos de los problemas encontrados. No lo sabemos en este momento. Si el enfoque selectivo mHTT funciona mejor y se tolera mejor, esto podría respaldar la suposición de que una supresión global de HTT normal no es bien tolerada en las personas, y esto afectará a muchos otros programas que apuntan a reducir las copias tanto mutantes como normales de la Gen HTT.
Programas de terapia génica en desarrollo clínico.
El siguiente programa más avanzado en la clínica en este momento, único's AMT130, es un agente administrado por vía viral, un microARN (miARN) que también se dirige a ambas copias del gen HTT, actualmente en estudios de Fase 2. Puedes ver un bonito vídeo que describe este enfoque. AQUÍ.
Esperamos que los datos del ensayo se publiquen a finales de 2024, aunque el ensayo tiene una duración de cinco años y comenzó en 2021. Se han inscrito un total de 26 pacientes en EE. UU. y Europa. Esta terapia con AAV (virus adenoasociado) es invasiva, requiere neurocirugía y se administra directamente en el caudado y el putamen, las dos estructuras de los ganglios basales del cerebro más afectadas en la EH. Los estudios preclínicos en roedores, primates no humanos y cerdos han demostrado que la terapia es bien tolerada hasta por 1 año. Debido a las propiedades del serotipo de AAV empleado (AAV-5), el agente terapéutico se distribuye ampliamente por todo el cerebro, transmitiéndose a través de las fibras cerebrales que atraviesan e inervan el cuerpo estriado. Veremos si esta terapia es bien tolerada a largo plazo, pero los datos presentados por UniQure no han mostrado evidencia de efectos nocivos hasta el momento en las dos dosis administradas (con varias cantidades del producto AAV-AMT130). Esto no debe darse por sentado, ya que este es el primer ensayo clínico de terapia génica AAV en la historia de la investigación de la EH. El entorno cerebral degenerado es un entorno complicado para terapias invasivas como ésta debido a un sistema inmunológico hiperactivo desencadenado por la muerte de las células cerebrales; ¡El hecho de que los pacientes puedan tolerar bien la cirugía y la terapia es un logro en sí mismo!
Recently, UniQure obtained from the FDA the Regenerative Medicine Advanced Therapy (RMAT) designation, laying the path for a Phase III trial in HD (see the press release AQUÍ). Como los estudios clínicos aún están en curso (incluido un grupo en el que los individuos reciben terapia con medicamentos inmunosupresores), debemos esperar hasta finales de 2024 para ver el paquete completo de datos de este importante ensayo.
Otros esfuerzos de terapia génica también emplean AAV, aunque cada empresa que busca estos agentes ha elegido diferentes tipos virales; En el caso de Viajero, eligieron AAV-1. El agente administrado a través de este virus también se dirige a ambas copias del gen HTT y, por lo tanto, será importante comparar sus efectos con los del programa Uniqure. Esperamos ver este programa en la clínica pronto.
Finalmente, la última empresa que esperábamos iniciar ensayos de terapia génica en EH con un agente selectivo de mHTT era Takeda, que estaba desarrollando un agente represor de dedos de zinc (ZFP) que disminuye selectivamente y de forma muy significativa la expresión de mHTT sin afectar la expresión normal de HTT. , también entregado por AAV-9. Había muchas expectativas sobre esta terapia, que fue desarrollada inicialmente por Sangamo Therapeutics, ya que es la única terapia selectiva de alelos para una amplia población en desarrollo. Desafortunadamente, Takeda anunció la interrupción de sus programas de terapia génica, incluido el programa ZFP para HD.

Fármacos orales de molécula pequeña que reducen el HTT
Los últimos agentes terapéuticos que han surgido son una nueva clase de agentes orales de molécula pequeña que se dirigen a la expresión de HTT en todo el cuerpo. Estos agentes se identificaron inicialmente en pantallas fenotípicas dirigidas a los niveles de expresión del gen de la neurona motora espinal (SMN)-2, como tratamiento para la atrofia muscular espinal (AME).
branalam (también llamado LIM070) es un fármaco de Novartis que actúa para aumentar los niveles de proteína SMN-2. Branaplam fue evaluado en una fase 2 para el tratamiento de la EH (el estudio VIBRANT-HD). El análisis de selectividad mostró que Branaplam puede disminuir la expresión de ambas copias de la proteína HTT mediante la modulación de empalme del ARNm de HTT. El mecanismo de acción de Branaplam parece conducir a la degradación del ARNm de HTT y a la disminución de los niveles de proteínas. En 2021, Novartis recibió la designación de medicamento huérfano de la FDA de EE. UU. para Branaplam en la EH, pero el estudio de fase 2b se detuvo debido a eventos adversos (se observó neuropatía periférica en algunos pacientes) y el programa se suspendió.
Otra empresa que busca moduladores de empalme para reducir el HTT es Terapéutica PTC. La empresa está realizando un ensayo de fase 2a (el estudio PIVOT-HD) con su fármaco. PTC-518 en sujetos con EH, y se espera que los resultados se anuncien más adelante en 2024. Esta misma semana, PTC Therapeutics anunció en un comunicado de prensa (ver AQUÍ) que PTC-518 fue bien tolerado y redujo los niveles de HTT (ambos alelos) en la periferia (midiendo HTT en las células sanguíneas) y el LCR (a la dosis más alta de 10 mg, informan una reducción de 43% en mHTT). Este es el primer fármaco oral de molécula pequeña con distribución en todo el cuerpo que muestra una reducción del HTT en todo el cuerpo en un estudio clínico. PTC también informa una desaceleración de los síntomas motores en pacientes que toman la dosis más alta de PTC-518.
La entrada de estos agentes en HD permite la posibilidad, por primera vez, de probar fármacos orales y de penetración cerebral para reducir la expresión de HTT en todo el cuerpo, lo que evita los problemas de distribución que se encuentran con los agentes de terapia génica. Sin embargo, queda por ver si la disminución sistémica crónica de ambos alelos del HTT El gen será bien tolerado en pacientes adultos y dará lugar a efectos modificadores de la enfermedad sostenidos.
El impacto de la genética humana en el nuevo desarrollo terapéutico
En los últimos años, se han identificado nuevos objetivos que parecen estar implicados en la lentitud o rapidez con la que una persona con la mutación avanza hasta una etapa clínica. La edad de inicio (AOO) en la EH se define como la fecha en la que un neurólogo clínico diagnostica que un individuo tiene síntomas motores típicos de la EH. Este "hito" en la progresión de la EH se eligió como un momento importante en el avance de la enfermedad para evaluar si las influencias genéticas pueden afectar la tasa de progresión. Desde hace muchos años se sabe que algunos individuos pueden desarrollar síntomas motores de la EH mucho antes o mucho más tarde que el "promedio" de individuos positivos para la EH con una mutación que presenta la misma longitud de repeticiones CAG en el gen HTT. Los grupos de investigación pudieron identificar aproximadamente una docena de genes asociados con este cambio en la tasa promedio de progresión. Este trabajo, en un gran estudio GWAS (estudio de asociación de todo el genoma) (consulte la última publicación AQUÍ), condujo a la identificación de genes implicados en la reparación del ADN y la expansión de las repeticiones CAG en las células somáticas (todas las demás células excepto las reproductivas del cuerpo). La comprensión mecanicista de cómo estos genes modifican potencialmente la progresión de la EH antes de que aparezcan los síntomas clínicos ha llevado a algunas empresas a intentar desarrollar terapias dirigidas a este mecanismo. El programa más avanzado hasta el momento se dirige a la expresión del gen MSH3 o su actividad, que se ha demostrado que modula la inestabilidad somática de la repetición HTT CAG. Ninguno de estos programas se encuentra todavía en desarrollo clínico, pero hay avances significativos hacia la obtención de nuevas terapias dirigidas a este mecanismo en la clínica.




