Acerca de la enfermedad de Huntington (EH)
La EH es una rara enfermedad neurodegenerativa autosómica dominante que generalmente ataca durante la mediana edad, entre los 35 y los 55 años, y se caracteriza por movimientos motores voluntarios e involuntarios, deterioro del comportamiento y deterioro cognitivo, lo que conduce a una dependencia completa y finalmente a la muerte después de un lento pero una progresión implacable a lo largo de dos décadas. Se estima que la prevalencia de HD manifiesta (sintomática) es de 5,7 por 100 000 habitantes en Europa, América del Norte y Australia, y de 0,4 por 100 000 en Asia. Compare esto con otras enfermedades neurodegenerativas comunes, que tienden a ser esporádicas y no causadas por mutaciones genéticas conocidas (por lo general, las mutaciones genéticas representan solo 2-10% de todos los casos de estos trastornos): Para la ELA, la prevalencia es de 2:100 000 personas ( 3 veces menos frecuente que HD); para la enfermedad de Parkinson, aproximadamente 1 millón de estadounidenses se ven afectados actualmente (aproximadamente 1 de cada 300 personas). El Alzheimer es la afección neurodegenerativa más común y afecta a más de 6 millones de estadounidenses. La enfermedad de Huntington es una de las más comunes genético trastornos cerebrales en todo el mundo, ya que todos los casos son causados por una sola mutación genética en un gen, el Huntingtin gene.
Cada descendiente de un individuo afectado de EH tiene una probabilidad 50% de heredar la mutación
En consecuencia, se cree que el tamaño de esta población 'en riesgo' definida como familiar de primer grado de un individuo afectado por la EH cuyo estado genético se desconoce es mayor, con estimaciones para la población occidental entre 30 y 44,9 por 100.000. Si bien existen pocos estudios que proporcionen datos de prevalencia de la EH en los conglomerados latinoamericanos, la prevalencia de individuos afectados por la EH en la región del Estado Zulia (Venezuela) se estima en 700 por 100.000, la más alta del mundo.
Historia de la investigación de la EH en Venezuela
La mutación del gen para la enfermedad de Huntington (EH) fue descubierta en 1993 por un consorcio de investigadores interdisciplinarios financiado por los NIH llamado Proyecto Colaborativo de Investigación de EE. muestras de sangre recolectadas de familias afectadas por la EH que viven en y alrededor del lago de Maracaibo en Venezuela, una región de aislamiento geográfico que resulta en una mayor prevalencia de la EH. Muchos hallazgos científicos y médicos que han tenido un gran impacto en nuestra comprensión de la enfermedad, incluida la identificación del gen causante, la evidencia de una ganancia tóxica de la función de la mutación a través del estudio de individuos homocigotos y la identificación de genes modificadores que alteran la edad de inicio de la EH (un área de renovado interés terapéutico), fue posible gracias a la amplia participación de estos pacientes venezolanos con EH, sus familias y sus comunidades.
En palabras de la Dra. Nancy Wexler, la científica pionera que dirigió el trabajo para identificar la mutación que causa la EH:
“Las familias venezolanas representan la población HD más grande y mejor caracterizada del mundo. Se han realizado veintitrés años de estudios prospectivos —genéticos, neurológicos y cognitivos— con afines […]. Esperamos que los venezolanos puedan beneficiarse plenamente de los resultados de la investigación, ya que desempeñaron un papel fundamental para hacer que todo fuera posible”.
Wexler, 2012
Hoy en día, esta comunidad sigue siendo uno de los grupos de HD más empobrecidos del mundo sin que se aborden sus necesidades humanas básicas, en gran parte debido a la devastadora situación política actual en Venezuela. Este fue un factor en nuestra decisión de iniciar Factor-H, una organización dedicada a brindar asistencia humanitaria y médica a estas poblaciones.
El gen que causa la enfermedad de Huntington
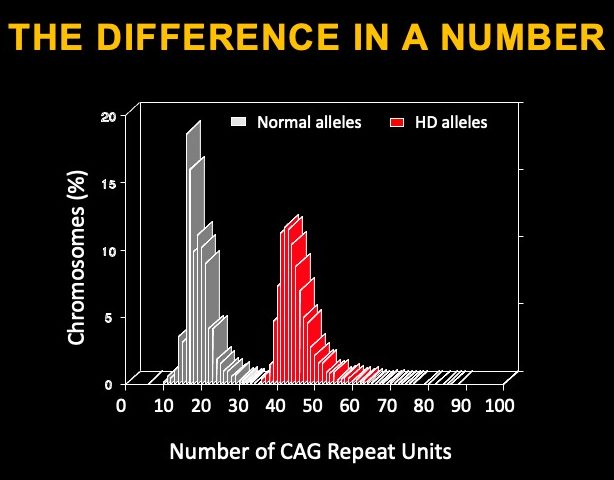
El gen afectado en la enfermedad de Huntington se llama Huntingtin, y es un gen muy grande. El gen codifica una proteína de 3144 aminoácidos con muchas funciones asignadas y que se cree que es esencial durante el desarrollo embrionario y para el funcionamiento del cerebro adulto. La mutación que causa la EH se encuentra dentro del comienzo del gen de la Huntingtina, en una región del ADN llamada 'exón 1', la región del ADN que codificó el comienzo de la proteína Huntingtina (HTT). Esta región de ADN incluye una repetición de secuencia que contiene citosina-adenina-guanina (CAG) que es muy variable entre los individuos; el rango típico es de 17 a 22 repeticiones. En los individuos con EH afectados, la longitud de la repetición en la copia mutada (o 'alelo') es superior a 39 repeticiones. Cuanto más larga sea la repetición, antes aparecerán los síntomas de la enfermedad. Las repeticiones CAG se traducen en tramos del aminoácido glutamina en la proteína.

Si bien aún no sabemos exactamente cómo la expansión de esta repetición conduce a la enfermedad, las repeticiones hacen que la proteína sea inestable. El análisis patológico en los cerebros de pacientes fallecidos muestra que la proteína HTT expandida se agrega y forma inclusiones intracelulares. La proteína HTT normal es una proteína que surgió temprano durante la evolución de los animales y está presente en los insectos y en todos los mamíferos. Hay muchas funciones asignadas a este gen, incluida la regulación de cómo se transportan varias vesículas a los lugares correctos, algo que es muy importante en el sistema nervioso, por ejemplo. También se cree que HTT está asociado con un conjunto de vesículas llamadas "fagosomas", o un tipo de vesículas que actúan como máquinas de reciclaje de la célula. Cuando se dañan, estos fagosomas no pueden reciclar el material celular y las células mueren. En el núcleo, mHTT puede acumularse y agregarse, dando lugar a una serie de problemas a nivel de expresión génica y regulación de la cromatina, y esto también puede dar lugar a problemas importantes.
Aún se debate mucho si la mutación la vuelve tóxica al afectar la función normal de la proteína HTT. Lo cierto es que, a pesar de 30 años de investigación, todavía no sabemos exactamente cómo funciona la HTT y qué funciones son críticas para la toxicidad impartida por la expansión polyCAG en el gen. Sin embargo, para mí está claro que HTT tiene muchas funciones y que la mutación puede alterar muchas de ellas. Lo que también está claro es que el gen HTT normal es esencial para la vida. Los embriones de roedores que carecen del gen no se desarrollan más allá del día 7-8 del desarrollo embrionario. Cuando los científicos eliminan el gen en los roedores adultos, dependiendo de cuándo se haga esto, se producen déficits en el cerebro, aunque aquí también hay controversia. No está claro por qué diferentes laboratorios llegan a diferentes conclusiones sobre el requisito de HTT normal en células cerebrales adultas, y probablemente se deba a problemas técnicos con la forma en que se realizan los experimentos, algo que esperamos aclarar pronto en CHDI. Sin embargo, mi veredicto es que la eliminación completa de ambas copias del gen HTT no es buena para el cerebro. Además de los estudios con roedores, los estudios con humanos también arrojaron luz sobre este tema: los laboratorios de Jim Gusella y Marcy Macdonald informaron sobre dos hembras que carecían de una copia del gen HTT (debido a la eliminación de un pequeño segmento del cromosoma 4 donde se encuentra el reside el gen HTT), y estos individuos están bien. Entonces puedes vivir con una copia del gen HTT. no tenemos células de esos individuos, por lo que no sabemos si expresan HTT a niveles de 50%. En otros estudios más recientes, se encontraron individuos con una expresión muy baja de HTT, debido a dos mutaciones diferentes que trabajaron para reducir la cantidad de proteína producida. Estos individuos heredaron mutaciones puntuales (un cambio en las bases individuales) de cada uno de sus padres: estos individuos padecían anomalías en el desarrollo, incluso en la función cerebral. Nuevamente, esto argumenta fuertemente que la HTT es necesaria para el desarrollo normal, y que los hallazgos en roedores también podrían aplicarse a los humanos, debido a la conservación de la función del gen HTT.
Independientemente de cómo la HTT mutante cause la EH, el área principal para el desarrollo terapéutico es eliminar o disminuir la expresión de la proteína HTT mutada. Los fundadores y miembros de la junta de Factor-H son científicos líderes que desarrollan tratamientos para la EH y esperan poner nuevas terapias a disposición de las comunidades a las que servimos. Para obtener más información sobre los programas terapéuticos actuales, consulte la sección de Noticias.




