From a gene mutation to a lethal disease
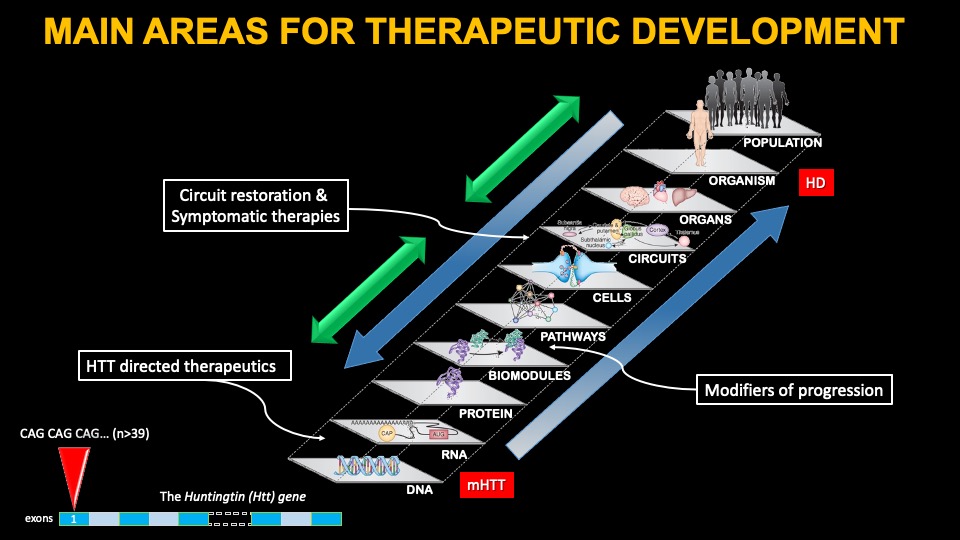
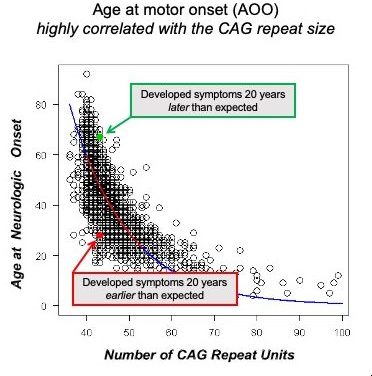
HD is caused by a single mutation in a region of the HTT gene, located at the beginning of the gene (in exon 1), which contains a repeat sequence comprised of CAG repeats; in the normal range, people have between 17-22 repeats on average; in HD individuals, an expansion greater than 39 leads inevitably to disease. The longer the number of repeats, typically, on average, the earlier the patients start showing visible symptoms of the disease.
This means that every person with HD has the same type of mutation in only one gene, making HD an unusual disorder. Most other common neurodegenerative diseases have many different origins, and only between 2-5% of all cases of Alzheimer’s, Parkinson’s or ALS have a genetic basis.
A lot of research has been dedicated to finding out how the mutation in the HTT gene leads to HD, and a lot of potential cellular mechanisms have been identified. The impact of the mutation has devastating consequences for many brain cells, and the theory goes that if we can understand how this happens, we might be able to develop therapies to treat the disease. However, these efforts have been largely unsuccessful thus far, including disappointing results with deep brain stimulation approaches. Some symptomatic therapies, targeting one or another mechanism thought to go awry in humans affected with HD are under development, and below we cover two of the most advanced therapies, Pridopidine and Valbenazine..
Another approach to treat HD is to target specific symptoms of the disease thought to arise from alterations in specific brain circuits. Mutant HTT leads to a loss of cells and brain fibers in specific circuits in the brain, which partially explain many of the symptoms of the disease. Therefore, strengthening or restoring those circuits might offer some benefit. In the case of HD, many circuits are affected, but the major ones involve the connection between various cortical areas and the basal ganglia, although other changes are known in the cerebellum and hypothalamus.
This is the ‘traditional’ approach taken for symptomatic therapies in every degenerative disorder, and one that has been effective in controlling some symptoms. For example, it led to the development of tetrabenazine for the control of chorea, and to the use of antipsychotic medications targeting the dopaminergic system for irritability, for example. However, at the moment, we lack therapies that target the most debilitating symptoms of HD – cognitive alterations and apathy.
News on symptomatic treatments
Pridopidine, formally known as ACR16, is being pursued by the company Prilenia. ACR16 was formerly developed by Teva. In a recently completed Phase III study (PIVOT-HD), Pridopidine failed to meet the primary endpoint of the trial but showed signs of improving some symptoms in the cognitive and motor domains only in the subset of HD patients not taking anti-dopaminergic medications such as tetrabenazine or antipsychotics. The primary endpoint was change from baseline to week 65 in TFC (the total functional capacity test). The key secondary endpoint was change from baseline in the composite Unified Huntington Disease Rating Scale (cUHDRS), and additional endpoints included quantitative motor (Q-Motor), the cognitive test Stroop Word Reading (SWR), and Quality of Life (HD-QoL). The Phase III study enrolled 499 individuals and was conducted in the U.S., Canada, Austria, Czech Republic, France, Germany, Italy, the Netherlands, Poland, Spain and the United Kingdom.
Prilenia is now appealing to regulatory authorities to enable a registration path for Pridopidine without having to conduct new clinical trials. Currently, they are awaiting feedback from the European Regulatory Agency (EMA) to see if their application for Market Authorization (MAA) is approved, which would enable the company to market the product in Europe.
Pridopidine has been investigated as a treatment for HD in three randomized, double-blind, placebo controlled clinical trials: HART, MermaiHD, and PRIDE-HD. Initial studies in HD focused on the effects of pridopidine in the motor endpoints, under the hypothesis that pridopidine had an effect on dopaminergic control of movement. Indeed, a small effect on motor endpoints was seen in the HART and MermaiHD studies, although people also saw a small effect on the total functional capacity scale (TFC) and extended the phase 2 study in an open-label modification to 52 weeks.
In the PRIDE-HD study, pridopidine dosing demonstrated a small but beneficial effect on TFC and this effect seemed to be more pronounced for early HD participants, which led the investigators to extend these observations in a Phase 3 study, PROOF-HD, currently enrolling and using the UHDRS-TFC score as the primary endpoint.
Valbenazine is a drug recently approved by the FDA (commercial name INGREZZA) after positive results on chorea in HD in the Phase III trial (KINECT-HD) developed by the company Neurocrine Biosciences. You can see the PDF of the news release HERE.
Key clinical trial outcomes from KINECT-HD include:
- A three-times greater improvement in chorea compared to placebo, from the start to the end of the 12-week clinical study. A reduction in chorea severity by about 40 percent from baseline to maintenance, and nearly half of patients saw a more than 40 percent reduction in chorea severity by Week 12.1
- Fifty-three percent of patients and 43 percent of healthcare professionals reported overall HD chorea symptoms were “very much improved” or “much improved”
Valbenazine works similarly to tetrabenazine but offers some advantages, namely once a day administration orally and a lesser adverse event profile. It remains to be seen how patients do when taking Valbenazine for long periods of time.
HTT-targeting gene therapies
By far the major area of investment to develop effective treatments for HD is targeting the expression of the Huntingtin gene. Ample research over many years in animal models of HD support the targeting of HTT expression as a disease modifying therapy. We know with certainty in mouse models that if one can decrease mutant HTT expression enough, manifestations can be minimized or altogether reversed. However, mouse models of HD lack significant features of the disease – most importantly, the death of brain neurons and an inflammatory environment. In addition, people with HD in the symptomatic stages have very extensive loss of neurons in several cortical areas and most nuclei of the basal ganglia, the regions most affected in HD. Therefore, we must be cautious assuming that the human HD brain will respond similarly to lowering HTT expression, given these very significant differences.
Most therapies currently under development target both copies of the HTT gene, both the mutated version and the normal (or ‘non-expanded’) copy. There is significant concern that lowering of the normal copy might lead to untoward effects after prolonged silencing. We know also from mouse studies that a complete loss of both copies of HTT is lethal during embryonic development. In the adult, the results are more mixed and significant loss (but not complete) of HTT can be better tolerated. In humans, we know that a few individuals have been found with only one copy of the HTT gene, and they do not exhibit the typical clinical manifestations of HD. However, individuals with mutations in both copies of the HTT gene and that show very low expression of HTT develop brain abnormalities during childhood. Therefore, lowering HTT too much is likely deleterious in humans as well.
ASO therapeutics in clinical development
This is a potential issue that might have derailed the Ionis/Roche Tominersen ASO program, which in Phase 3 clinical trials was stopped due to adverse events. In this trial, which generated a lot of expectations and hope for a first disease-modifying therapy in HD, the subjects exposed to the highest dose of Tominersen (an antisense oligonucleotide or ASO) exhibited a worsening in most clinical measures of HD, enlarged brain ventricles, and elevation of measures of toxicity and inflammation as judged in CSF collection analysis, leading to the trial being terminated prematurely. As of 2024, Roche is recruiting for a new Phase II study with Tominersen based on a post-hoc analysis of the data collected in the Phase III study, which indicated that HD affected individuals with low CAP scores (a measure of disease progression based on age and CAG repeat length) and young in age tolerated the drug more. You can read Roche’s rationale in this NEJM article. In addition to recruiting younger and less affected patients, the study will be conducted with a lower dose of tominersen, which can impact the distribution of the drug to deeper brain areas affected in HD.
Around the same time of the Tominersen failure, in March 2021, two other ASO programs being developed by Wave Life Sciences, were also stopped, this time in Phase 1/2, due to a lack of pharmacological effects of the expression of mHTT in the CSF of dosed patients. Compared to the Roche trial with Tominersen, the Wave ASOs failed to lower mHTT enough in the CSF, leading to the company stopping their development. However, the company persevered and developed a third ASO targeting a single nucleotide polymorphism (SNP) found in a subset of HD patients of European descent, named WVE-003. This ASO, contrary to the Roche ASO Tominersen, is selective for the mutant copy of the HTT gene. Last week, Wave announced positive results of their WVE-003 molecule in a Phase 1/2 study (SELECT-HD trial), demonstrating safety and a significant reduction in mHTT in patient CSF (45% lowering). In addition, there seems to be a stabilization of caudate volume and the total functional score. You can read the press release HERE.
This is indeed very exciting news, as this is the first instance that a mHTT-selective therapy has shown an effect in lowering mHTT in HD patients. The trial also showed that the WVE-003 therapy was well tolerated and did not report increased ventricular volumes such as those commonly observed with Tominersen. We are awaiting the public disclosure of the trial results, including safety and clinical data on key functional and motor endpoints.
It is difficult to know why Tominersen led to the unfortunate events uncovered in the Ionis/Roche trial – it could be explained by a loss of normal HTT function due to excessive lowering in some parts of the spinal cord or the brain cortex (the areas most targeted by these spinal cord infusions); but it could also be a consequence of the agent employed: antisense oligonucleotides can be pro-inflammatory, and their accumulation after repeated dosing could also explain some of the issues encountered. We do not know at this point. If the mHTT selective approach works better and is better tolerated, this might lend support to the assumption that a global suppression of normal HTT is not well tolerated in people, and this will affect many other programs aiming to lower both mutant and normal copies of the HTT gene.
Gene therapy programs in clinical development
The next most advanced program in the clinic at the moment, Uniqure‘s AMT130, is a virally delivered agent, a microRNA (miRNA) also targeting both copies of the HTT gene, currently in Phase 2 studies. You can see a nice video describing this approach HERE.
We expect a trial data release at the end of 2024, even though the trial has a duration of five years and commenced in 2021. A total of 26 patients have been enrolled in the USA and Europe. This AAV (adeno-associated virus) therapy is invasive, requires neurosurgery, and is administered directly into the caudate and putamen, the two structures of the basal ganglia of the brain most affected in HD. Preclinical studies in rodents, non-human primates and pigs have shown the therapy to be well-tolerated up to 1 year. Due to the properties of the AAV serotype employed (AAV-5), the therapeutic agent distributes broadly throughout the brain, being transmitted via the brain fibers that pass through and innervate the striatum. We will see whether this therapy is well tolerated in the long term, but data presented by UniQure has shown no evidence of deleterious effects thus far in the two doses administered (with various quantities of the AAV-AMT130 product). This is not to be taken for granted, being that this is the first gene therapy AAV clinical trial in the history of HD research. The degenerating brain environment is a complicated environment for invasive therapies such as this due to a hyperactive immune system triggered by the death of brain cells; the fact that patients can tolerate the surgery and the therapy well is an achievement in itself!
Recently, UniQure obtained from the FDA the Regenerative Medicine Advanced Therapy (RMAT) designation, laying the path for a Phase III trial in HD (see the press release HERE). As the clinical studies are still ongoing (including an arm where individuals receive the therapy with immunosuppressive drugs), we need to wait until the end of 2024 to see the entire data package from this important trial.
Other gene therapy efforts also employ AAVs, albeit each company pursuing these agents have chosen different viral types; in the case of Voyager, they chose AAV-1. The agent delivered via this virus, also targets both copies of the HTT gene, and therefore will be important to compare its effects to those of the Uniqure program. We hope to see this program in the clinic soon.
Finally, the last company which we hoped to start gene therapy trials in HD with a mHTT-selective agent was Takeda, who was developing a zinc-finger repressor (ZFP) agent that selectively decreases very significantly the expression of mHTT without affecting normal HTT expression, also being delivered by AAV-9. There was a lot of expectation about this therapy, which was initially developed by Sangamo Therapeutics, as this is the only broad-population allele-selective therapy in development. Unfortunately, Takeda announced a discontinuation of their gene therapy programs, including the ZFP program for HD.

Small molecule oral drugs lowering HTT
The latest therapeutic agents to emerge are a new class of oral small molecule agents targeting HTT expression throughout the body. These agents were initially identified in phenotypic screens targeting expression levels of the spinal motor neuron (SMN)-2 gene, as a treatment for spinal muscular atrophy (SMA).
Branaplam (also called LIM070) is a Novartis drug which acts to increase levels of SMN-2 protein. Branaplam was evaluated in a phase 2 for the treatment of HD (the VIBRANT-HD study). Selectivity analysis showed that Branaplam can decrease the expression of both copies of the HTT protein via splicing modulation of the HTT mRNA. The mechanism of action of Branaplam seems to be led to the decay of the HTT mRNA and decreasing protein levels. In 2021, Novartis received US FDA Orphan Drug Designation for Branaplam in HD, but the Phase 2b study was stopped due to adverse events (peripheral neuropathy in some patients was observed), and the program was discontinued.
Another company pursuing splicing modulators to lower HTT is PTC therapeutics. The company is conducting a Phase 2a trial (the PIVOT-HD study) with their drug PTC-518 in HD subjects, expecting results to be announced later in 2024. Just this week, PTC Therapeutics announced in a press release (see HERE) that PTC-518 was well tolerated and lowered HTT levels (both alleles) in the periphery (measuring HTT in blood cells) and the CSF (at the highest dose of 10 mgs, they report a 43% reduction in mHTT). This is the first small molecule oral drug with entire body distribution that shows a lowering of HTT throughout the body in a clinical study. PTC also reports a slowing of motor symptoms in patients taking the highest dose of PTC-518.
The entry of these agents in HD enables the possibility, for the first time, to test oral and brain penetrant drugs to lower HTT expression throughout the body, which circumvents the distribution issues encountered with gene therapy agents. However, it remains to be seen if the chronic, systemic lowering of both alleles of the HTT gene will be well tolerated in adult patients and if they will lead to sustained, disease modifying effects.
The impact of human genetics on new therapeutic development
In the last couple of years, new targets have been identified that seem to be implicated in how slow or how fast a person with the mutation advances to a clinical stage. The age-of-onset (AOO) in HD is defined as the date where a clinical neurologist diagnoses an individual with having motor symptoms typical of HD. This ‘milestone’ in the progression of HD was chosen as an important time in the disease advancement to evaluate whether genetic influences can affect the rate of progression. It was well known for many years that some individuals can develop motor symptoms of HD much earlier or much later than the ‘average’ of HD positive individuals with a mutation bearing the same length of CAG repeats in the HTT gene. The research groups were able to identify a dozen or so genes that were associated with this change in the average rate of progression. This work, in a large GWAS (genome-wide association study) study (see the latest publication HERE), led to the identification of genes implicated in DNA repair and the expansion of the CAG repeats in somatic cells (all other cells besides reproductive cells of the body). The mechanistic understanding of how potentially these genes modify the progression of HD prior to clinical symptoms has led to some companies trying to develop therapies targeting this mechanism. The most advanced program thus far targets the expression of the MSH3 gene or its activity, which has been shown to modulate somatic instability of the HTT CAG repeat. None of these programs is in clinical development yet, but there is significant progress towards getting new therapies targeting this mechanism in the clinic.




