
Sandrine Humbert from INSERM, Grenoble Institute for Neurosciences, spoke about the role of wild type HTT in brain development. Sandrine postulates that a significant component of HD pathogenesis involves the loss of function of normal HTT function(s), which involve, among other roles, the assembly of molecular complexes, including those transported by microtubules, and in the context of ciliogenesis. She also speculates that a primary pathological event is the loss of cortical afferent support for striatal spiny production neurons, partly by a loss of neurotrophic support driven by BDNF/TrkB signaling. Sandrine reviewed the prior work supporting these hypotheses, which lend credence to the concept that the developmental functions of HTT are impacted in HD. A major effect of the loss of normal HTT during development is alterations on cell cycle progression during neurogenesis (less neurons being generated) and changes in neuronal polarity, which get reflected in many different contexts during the development of cortical neurons, including in dendritic morphology in postmitotic neurons.
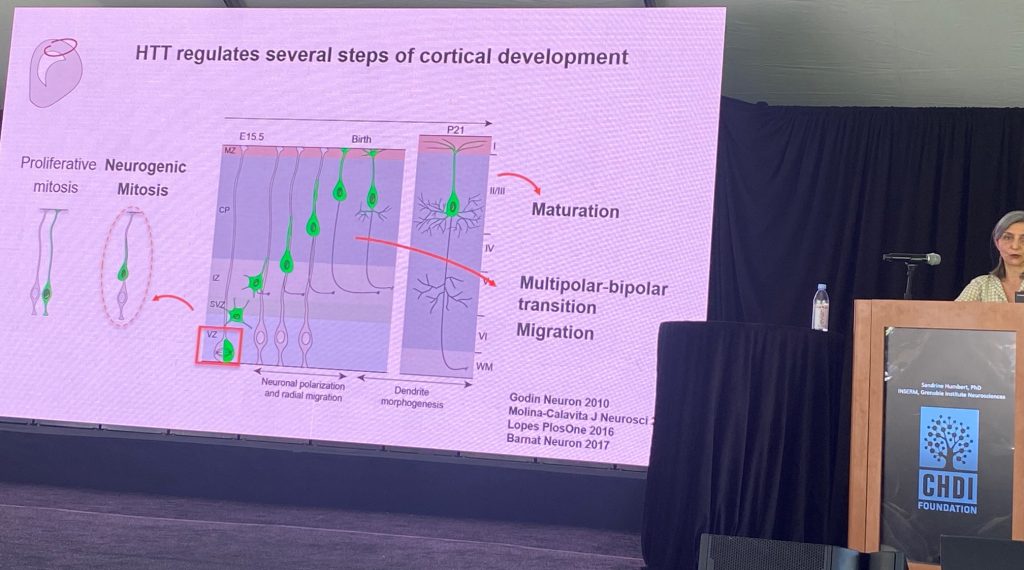
Some of the deficits observed in the rodent HTT knockouts seem to be found as well in histological analysis conducted in human fetal tissues, albeit the sample size is small. There seems to be converging evidence of a change in HTT localization in HD and a change in cellular polarity early in development of the cortex, as well as in deficits in the number and morphology of cilia in cortical progenitor cells. Other apparently shared observations between rodent models and HD individuals include partial agenesis of the corpus callosum (that is, that some of the well known loss of callosal fibers in HD are due to a problem of axonal growth during development). Sandrine then described the role of the protein NUMA1 in axonal growth cone deficits in neurons derived from knock-in mouse models. The over expression of NUMA1 in Q111 mice can restore the callosal developmental deficits in this model. They also showed that epothiloneB, a natural product that interferes with tubulin polymerization and also leads to improvements in callosal structure in this model.
RAFAELLE LENNACO
Rafaelle Iennaco (postdoc of Dr. Elena Cattaneo’s group), University of Milano spoke about the role of specific residues within exon-1 of the HTT gene in modulating its function. Rafaelle has engineered mouse stem cells expressing various deletions and point mutations in the HTT gene, and their relationship to its developmental function. In evolution, the number of CAG repeats starts to increase from birds, reptiles, fishes and amphibians (4Qs), whereas in mammals the range is between 5-35 CAG repeats. The N17 is very conserved, but the poly-proline domain (the ‘PRD’) is highly divergent too across evolution, which only appears in mammals. However, the role of these 3 regions and the importance of specific residues has not been slides in sufficient depth. Therefore, they engineered a large set of mES cells expressing various length of the polyCAG tract (Q= 0, 2, 4, 7 ,17 etc.) and shown to impact an early developmental defect observed in culture. they also identified a set of genes whose expression correlated with the number of CAGs in the normal range, revealing genes implicated in nervous system development.
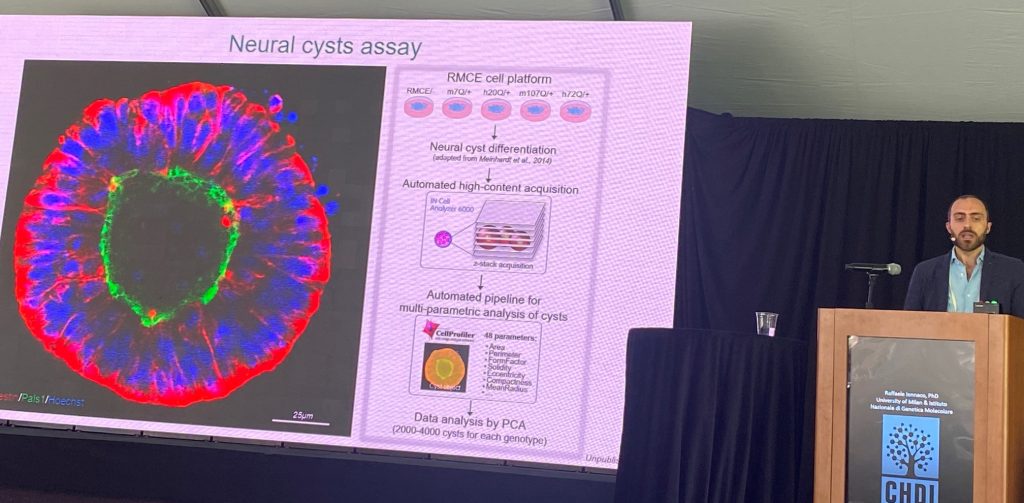
The next stage of the presentation focused on understanding the pathogenic role of the human mutant HTT (more pathogenic than the mouse exon 1). They compared the same pathogenic ranges of CAG repeats in the human context versus the mouse context, in the context of the morphology of neuronal ‘cysts’ and in dendritic morphologies in culture, supporting that the human sequences matter in triggering these deficits. Since the main changes between exon-1 of mouse vs human HTT resides in the polyproline region, a set of knock-in stem cells was generated where the polypro regions where switched between human and mouse. the human polyproline region is important for pathogenesis in these two systems. Human region worsens, and mouse region ameliorates, the described phenotypes.
DR. BALJIT KHAKI
Dr. Baljit Khaki from UCLA described his work on the pathogenic impact of mHTT in R6/2 and Q175 mouse models, in terms of cell-specific deficits, by comparing the pathogenic role of mHTT in astrocytes vs neurons in the striatum (at a physiological, molecular and behavioral level). Mal is a world leader of astrocyte physiology and their role in normal brain functions and in disease. In this seminar, Bal showed that many of these deficits can be prevented/restored after striatal cells are exposed to a mHTT specific zinc finger protein repressor (ZFP) that inhibits expression of mHTT specifically without affecting normal HTT expression. The novelty of his recent work is the fact that his team expressed the mHTT ZFP only in neurons or only in astrocytes, and is able to analyze the impact of suppressing mHTT in terms of cell-autonomous vs non-cell autonomous mechanisms – therefore pointing to the cell type most important in driving pathogenic mechanisms. That is, lower mHTT in astrocytes and study effects on astrocytes and in neurons (which express mHTT still), and vice versa. His results overwhelmingly support that, in mouse HD models, the neuronal expression of mHTT is the most significant event in driving disease progression, and that most of the astrocyte changes are adaptive to the deficits triggered in striatal neurons by mHTT.

Bal also describes the fact that astrocytic GPCRs that signal via Gi coupling can partially restore disease symptoms in various HD models, demonstrating that this is a potentially valuable therapeutic strategy. He expanded these earlier efforts, published in 2021, to study the role of mHTT suppression effects in neurons vs astrocytes to understand the mechanisms by which mHTT contributes to disease and the cell types most important for disease. The ZFPs were expressed in AAV viruses and used extensively in vivo at various stages of disease development. A key message is that changes in astrocytes relate to cholesterol metabolism are confirmed, and that this pathway can be rescued cell-autonomously when mHTT is suppressed in astrocytes. Bal’s lab also used electrophysiological techniques to show restoration of abnormal firing and cell properties after neuronal, but not astrocytic, mHTT lowering.

A final set of experiments was to lower mHTT throughout the brains by the use of AAV PHPeb to drive expression of the ZFP. This treatment was shown to ameliorate disease measures in R6.2 mice, a very aggressive model of HD. These studies also showed that neuronal expression of ZFP but not astrocytic. expression rescues the behavioral deficits by multiple endpoints (nesting, open field, clasping, etc.).


