About Huntington’s disease (HD)
HD is a rare autosomal dominant neurodegenerative disease that typically strikes during mid-life, between 35-55 years old, and is characterized by voluntary and involuntary motor movements, behavioral impairment and cognitive decline, leading to complete dependence, and ultimate death after a slow but unrelenting progression over two decades. The prevalence of manifest (symptomatic) HD is estimated to be 5.7 per 100,000 of the population in Europe, North America, and Australia, and 0.4 per 100,000 in Asia. Compare this to other common neurodegenerative diseases, which tend to be sporadic and not caused by known genetic mutations (typically, genetic mutations account for only 2-10% of all cases for these disorders): For ALS, the prevalence is 2:100,000 people (3 times less frequent than HD); for Parkinson’s disease, approximately 1 million Americans are affected currently (roughly 1 in 300 people). Alzheimer’s is the most common neurodegenerative condition, affecting more than 6 million Americans. Huntington’s disease is one of the most common genetic brain disorders worldwide, as all cases are caused by a single genetic mutation in one gene, the Huntingtin gene.
Each offspring of an affected HD individual has a 50% chance of inheriting the mutation
Consequently, the size of this ‘at risk’ population defined as a first-degree relative of an HD-affected individual whose genetic status is unknown is thought to be higher, with estimates for the Western population between 30 and 44.9 per 100,000. While there are few studies providing prevalence data of HD in Latin American clusters, the prevalence of HD-affected individuals in the region of Zulia State (Venezuela) is estimated to be 700 per 100,000, the highest in the world.
History of HD research in Venezuela
The gene mutation for Huntington’s disease (HD) was discovered in 1993 by an NIH-funded research consortium of interdisciplinary investigators called the U.S.-Venezuela Research Collaborative Project that mapped the genome through linkage analysis in 1983, then later isolated the gene, using hundreds of blood samples collected from HD-affected families living on and around Lake Maracaibo in Venezuela, a region of geographical isolation resulting in a higher prevalence of HD. Many scientific and medical findings that have greatly impacted our understanding of the disease, including the identification of the causative gene, the evidence for a toxic gain of function of the mutation through the study of homozygote individuals, and the identification of modifier genes that alter the age of onset of HD (an area of renewed therapeutic interest), was made possible through the extensive participation of these Venezuelan HD patients, their families and their communities.
In the words of Dr. Nancy Wexler, the pioneer scientist who led the work to identify the mutation that causes HD:
“The Venezuelan kindreds represent the largest and best characterized HD population in the world. Twenty-three years of prospective studies—genetic, neurological, and cognitive—have been carried out with kindred members […]. We hope that the Venezuelans will be able to benefit fully from the results of the research, as they played such a critical role in making everything possible.”
Wexler, 2012
Today this community remains one of the most impoverished HD clusters in the world without their basic human needs being addressed, in large part due to the current devastating political situation in Venezuela. This was a factor in our decision to start Factor-H, an organization dedicated to providing humanitarian and medical assistance to these populations.
The gene that causes Huntington’s disease
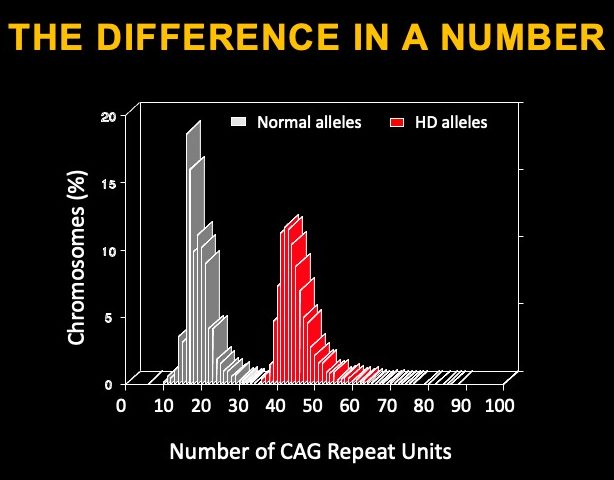
The gene affected in Huntington’s disease is called Huntingtin, and it is a very large gene. The gene codes for a protein of 3144 amino acids with many functions assigned to it and thought to be essential during embryonic development and for adult brain function. The mutation that causes HD is located within the beginning of the Huntingtin gene, in a region of DNA called ‘exon 1’ – the region of DNA that codified the beginning of the Huntingtin (HTT) protein. This DNA region includes a sequence repeat containing cytosine-adenine-guanine (CAG) that is highly variable across individuals – the typical range is 17-22 repeats. In affected HD individuals, the repeat length in the mutated copy (or ‘allele’) is greater than 39 repeats. The longer the repeat, the earlier the disease symptoms appear. The CAG repeats are translated into stretches of the amino acid glutamine in the protein.

While we don’t yet know exactly how the expansion of this repeat leads to disease, the repeats make the protein unstable. Pathological analysis in the brains of deceased patients show that the expanded HTT protein aggregates and forms intracellular inclusions. The normal HTT protein is a protein that arose early during the evolution of animals, and is present in insects and all mammals. There are many functions assigned to this gene, including the regulation of how various vesicles get transported to the right places – something that is very important in the nervous system, for example. HTT is also thought to be associated with a set of vesicles called ‘phagosomes’, or a type of vesicles that act as the recycling machines of the cell. When damaged, these phagosomes are unable to recycle cellular material, and cells die. In the nucleus, mHTT can accumulate and aggregate, leading to a host of problems at the level of gene expression and regulation of the chromatin, and this can also lead to significant problems.
Whether the mutation renders it toxicity by affecting the normal function of the HTT protein is still very much under discussion. The truth is, spite 30 years of research, we still do not know exactly how HTT works and which functions are critical for the toxicity imparted by the polyCAG expansion in the gene. It is clear to me, however, that HTT has many functions, and that the mutation can alter many of them. What is also clear is that the normal HTT gene is essential for life. Rodent embryos that lack the gene do not develop past day 7-8 of embryonic development. When scientists delete the gene in the adult rodents, depending on when this is done – deficits in the brain ensue, although here too there is controversy. Why different labs come to different conclusions about the requirement of normal HTT in adult brains cells is unclear, and prob ably due to technical issues with how the experiments are done, something we hope to clarify at CHDI soon. However, my verdict is that the complete elimination of both copies of the HTT gene is not good for the brain. Aside from rodent studies, human studies have also shed light onto this topic: the labs of Jim Gusella and Marcy Macdonald have reported on two females who lacked one copy of the HTT gene (due to a deletion of a small segment of chromosome 4 where the HTT gene resides), and these individuals are fine. So you can live with one copy of the HTT gene. we don’t have cells from those individuals so we do not know if they express HTT at 50% levels. In other, more recent studies, individuals were found with very low expression of HTT – due to two different mutations that worked to lower the amount of the protein produced. These individuals inherited point mutations (a change in single bases) from each of their parents: these individuals suffered from developmental abnormalities, including in brain function. Again, this strongly argues that HTT is required for normal development, and that the findings in rodents might very well apply to humans as well, due to the conservation of function of the HTT gene.
Regardless of how mutant HTT causes HD – the major area for therapeutic development is eliminating or decreasing the expression of the mutated HTT protein. The Factor-H founders and board members are leading scientists developing treatments for HD, and hope to make new therapies available to the communities we serve. To find out more about current therapeutic programs, see the News section.





